From Wikipedia, the free encyclopedia
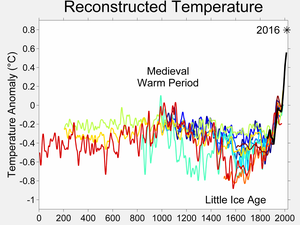
The reconstructed depth of the Little Ice Age varies between different
studies (anomalies shown are from the 1950–80 reference period)
The Little Ice Age (LIA) was a period of cooling that occurred after the Medieval Warm Period.[1] Although it was not a true ice age, the term was introduced into scientific literature by François E. Matthes in 1939.[2] It has been conventionally defined as a period extending from the 16th to the 19th centuries,[3][4][5] but some experts prefer an alternative timespan from about 1300[6] to about 1850.[7][8][9] Climatologists and historians working with local records no longer expect to agree on either the start or end dates of the period, which varied according to local conditions.
The NASA Earth Observatory notes three particularly cold intervals: one beginning about 1650, another about 1770, and the last in 1850, all separated by intervals of slight warming.[5] The Intergovernmental Panel on Climate Change Third Assessment Report considered the timing and areas affected by the Little Ice Age suggested largely-independent regional climate changes rather than a globally-synchronous increased glaciation. At most, there was modest cooling of the Northern Hemisphere during the period.[10]
Several causes have been proposed: cyclical lows in solar radiation, heightened volcanic activity, changes in the ocean circulation, an inherent variability in global climate, or decreases in the human population.
Areas involved
The Intergovernmental Panel on Climate Change Third Assessment Report (TAR) of 2001 described the areas affected:Evidence from mountain glaciers does suggest increased glaciation in a number of widely spread regions outside Europe prior to the twentieth century, including Alaska, New Zealand and Patagonia. However, the timing of maximum glacial advances in these regions differs considerably, suggesting that they may represent largely independent regional climate changes, not a globally-synchronous increased glaciation. Thus current evidence does not support globally synchronous periods of anomalous cold or warmth over this interval, and the conventional terms of "Little Ice Age" and "Medieval Warm Period" appear to have limited utility in describing trends in hemispheric or global mean temperature changes in past centuries.... [Viewed] hemispherically, the "Little Ice Age" can only be considered as a modest cooling of the Northern Hemisphere during this period of less than 1°C relative to late twentieth century levels.[10]The IPCC Fourth Assessment Report (AR4) of 2007 discusses more recent research, giving particular attention to the Medieval Warm Period. It states that "when viewed together, the currently available reconstructions indicate generally greater variability in centennial time scale trends over the last 1 kyr than was apparent in the TAR.... The result is a picture of relatively cool conditions in the seventeenth and early nineteenth centuries and warmth in the eleventh and early fifteenth centuries, but the warmest conditions are apparent in the twentieth century. Given that the confidence levels surrounding all of the reconstructions are wide, virtually all reconstructions are effectively encompassed within the uncertainty previously indicated in the TAR. The major differences between the various proxy reconstructions relate to the magnitude of past cool excursions, principally during the twelfth to fourteenth, seventeenth and nineteenth centuries."[11]
Dating

The last written records of the Norse Greenlanders are from a 1408 marriage at Hvalsey Church, now the best-preserved of the Norse ruins.
There is no consensus regarding the time when the Little Ice Age began,[12][13] but a series of events before the known climatic minima has often been referenced. In the 13th century, pack ice began advancing southwards in the North Atlantic, as did glaciers in Greenland. Anecdotal evidence suggests expanding glaciers almost worldwide. Based on radiocarbon dating of roughly 150 samples of dead plant material with roots intact, collected from beneath ice caps on Baffin Island and Iceland, Miller et al. (2012)[6] state that cold summers and ice growth began abruptly between 1275 and 1300, followed by "a substantial intensification" from 1430 to 1455.[14]
In contrast, a climate reconstruction based on glacial length[15][16] shows no great variation from 1600 to 1850 but strong retreat thereafter.
Therefore, any of several dates ranging over 400 years may indicate the beginning of the Little Ice Age:
- 1250 for when Atlantic pack ice began to grow
- 1275 to 1300 based on the radiocarbon dating of plants killed by glaciation
- 1300 for when warm summers stopped being dependable in Northern Europe
- 1315 for the rains and Great Famine of 1315–1317
- 1550 for theorized beginning of worldwide glacial expansion
- 1650 for the first climatic minimum.
Northern Hemisphere
Europe

The Frozen Thames, 1677
The Little Ice Age brought colder winters to parts of Europe and North America. Farms and villages in the Swiss Alps were destroyed by encroaching glaciers during the mid-17th century.[20] Canals and rivers in Great Britain and the Netherlands were frequently frozen deeply enough to support ice skating and winter festivals.[20] The first River Thames frost fair was in 1607 and the last in 1814; changes to the bridges and the addition of the Thames Embankment affected the river flow and depth, greatly diminishing the possibility of further freezes. Freezing of the Golden Horn and the southern section of the Bosphorus took place in 1622. In 1658, a Swedish army marched across the Great Belt to Denmark to attack Copenhagen. The winter of 1794–1795 was particularly harsh: the French invasion army under Pichegru was able to march on the frozen rivers of the Netherlands, and the Dutch fleet was fixed in the ice in Den Helder harbour.
Sea ice surrounding Iceland extended for miles in every direction, closing harbors to shipping. The population of Iceland fell by half, but that may have been caused by skeletal fluorosis after the eruption of Laki in 1783.[21] Iceland also suffered failures of cereal crops and people moved away from a grain-based diet.[22] The Norse colonies in Greenland starved and vanished by the early 15th century, as crops failed and livestock could not be maintained through increasingly harsh winters, but Jared Diamond has suggested they had exceeded the agricultural carrying capacity before then. Greenland was largely cut off by ice from 1410 to the 1720s.[23]

Winter skating on the main canal of Pompenburg, Rotterdam in 1825, shortly before the minimum, by Bartholomeus Johannes van Hove
The Twentieth Century climatologist Hubert Lamb said that in many years, "snowfall was much heavier than recorded before or since, and the snow lay on the ground for many months longer than it does today."[24] In Lisbon, Portugal, snowstorms were much more frequent than today; one winter in the 17th century produced eight snowstorms.[25] Many springs and summers were cold and wet but with great variability between years and groups of years. Crop practices throughout Europe had to be altered to adapt to the shortened, less reliable growing season, and there were many years of dearth and famine (such as the Great Famine of 1315–1317, but that may have been before the Little Ice Age).[26] According to Elizabeth Ewan and Janay Nugent, "Famines in France 1693–94, Norway 1695–96 and Sweden 1696–97 claimed roughly 10 percent of the population of each country. In Estonia and Finland in 1696–97, losses have been estimated at a fifth and a third of the national populations, respectively."[27] Viticulture disappeared from some northern regions and storms caused serious flooding and loss of life. Some of them resulted in permanent loss of large areas of land from the Danish, German, and Dutch coasts.[24]
The violin maker Antonio Stradivari produced his instruments during the Little Ice Age. The colder climate is proposed to have caused the wood used in his violins to be denser than in warmer periods, contributing to the tone of his instruments.[28] According to the science historian James Burke, the period inspired such novelties in everyday life as the widespread use of buttons and button-holes, knitting of custom-made undergarments to better cover and insulate the body. Fireplace hoods were installed to make more efficient use of fires for indoor heating, and enclosed stoves were developed, with early versions often covered with ceramic tiles.[29]
The Little Ice Age, by anthropology professor Brian Fagan of the University of California at Santa Barbara, tells of the plight of European peasants during the 1300 to 1850 chill: famines, hypothermia, bread riots and the rise of despotic leaders brutalizing an increasingly dispirited peasantry. In the late 17th century, agriculture had dropped off dramatically: "Alpine villagers lived on bread made from ground nutshells mixed with barley and oat flour." [30] Historian Wolfgang Behringer has linked intensive witch-hunting episodes in Europe to agricultural failures during the Little Ice Age.[31]
Depictions of winter in European painting

The Reverend Robert Walker Skating on Duddingston Loch, attributed to Henry Raeburn, 1790s
William James Burroughs analyses the depiction of winter in paintings, as does Hans Neuberger.[32] Burroughs asserts that it occurred almost entirely from 1565 to 1665 and was associated with the climatic decline from 1550 onwards. Burroughs claims that there had been almost no depictions of winter in art, and he "hypothesizes that the unusually harsh winter of 1565 inspired great artists to depict highly original images and that the decline in such paintings was a combination of the 'theme' having been fully explored and mild winters interrupting the flow of painting".[33] Wintry scenes, which entail technical difficulties in painting, have been regularly and well handled since the early 15th century by artists in illuminated manuscript cycles showing the Labours of the Months, typically placed on the calendar pages of books of hours. January and February are typically shown as snowy, as in February in the famous cycle in the Les Très Riches Heures du duc de Berry, painted 1412–1416 and illustrated below. Since landscape painting had not yet developed as independent genre in art, the absence of other winter scenes is not remarkable.

The Hunters in the Snow by Pieter Brueghel the Elder, 1565
The famous winter landscape paintings by Pieter Brueghel the Elder, such as The Hunters in the Snow, are all thought to have been painted in 1565. His son Pieter Brueghel the Younger (1564–1638) also painted many snowy landscapes, but according to Burroughs, he "slavishly copied his father's designs. The derivative nature of so much of this work makes it difficult to draw any definite conclusions about the influence of the winters between 1570 and 1600...".[33][34]

Winter landscape with iceskaters, c. 1608, Hendrick Avercamp
Neuberger analysed 12,000 paintings, held in American and European museums and dated between 1400 and 1967, for cloudiness and darkness.[32] His 1970 publication shows an increase in such depictions that corresponds to the Little Ice Age,[32] peaking between 1600 and 1649.[35]
Paintings and contemporary records in Scotland demonstrate that curling and ice skating were popular outdoor winter sports, with curling dating back to the 16th century and becoming widely popular in the mid-19th century.[36] As an example, an outdoor curling pond constructed in Gourock in the 1860s remained in use for almost a century, but increasing use of indoor facilities, problems of vandalism, and milder winters led to the pond being abandoned in 1963.[37]
North America

"February" from the calendar of Les Très Riches Heures du duc de Berry, 1412–1416
Early European explorers and settlers of North America reported exceptionally severe winters. For example, according to Lamb, Samuel Champlain reported bearing ice along the shores of Lake Superior in June 1608. Both Europeans and indigenous peoples suffered excess mortality in Maine during the winter of 1607–1608, and extreme frost was reported in the Jamestown, Virginia, settlement at the same time.[24] Native Americans formed leagues in response to food shortages.[23] The journal of Pierre de Troyes, Chevalier de Troyes, who led an expedition to James Bay in 1686, recorded that the bay was still littered with so much floating ice that he could hide behind it in his canoe on 1 July.[38] In the winter of 1780, New York Harbor froze, allowing people to walk from Manhattan Island to Staten Island.
The extent of mountain glaciers had been mapped by the late 19th century. In the north and the south temperate zones, snowlines (the boundaries separating zones of net accumulation from those of net ablation) were about 100 metres (330 ft) lower than they were in 1975.[39] In Glacier National Park, the last episode of glacier advance came in the late 18th and the early 19th centuries.[40] In Chesapeake Bay, Maryland, large temperature excursions were possibly related to changes in the strength of North Atlantic thermohaline circulation.[41]
Mesoamerica
An analysis of several proxies undertaken in Mexico's Yucatan Peninsula, linked by its authors to Maya and Aztec chronicles relating periods of cold and drought, supports the existence of the Little Ice Age in the region.[42]Atlantic Ocean
In the North Atlantic, sediments accumulated since the end of the last ice age, nearly 12,000 years ago, show regular increases in the amount of coarse sediment grains deposited from icebergs melting in the now open ocean, indicating a series of 1–2 °C (2–4 °F) cooling events recurring every 1,500 years or so.[43] The most recent of these cooling events was the Little Ice Age. These same cooling events are detected in sediments accumulating off Africa, but the cooling events appear to be larger, ranging between 3–8 °C (6–14 °F).[44]Asia
Although the original designation of a Little Ice Age referred to reduced temperature of Europe and North America, there is some evidence of extended periods of cooling outside this region, but it is not clear whether they are related or independent events. Mann states:[3]While there is evidence that many other regions outside Europe exhibited periods of cooler conditions, expanded glaciation, and significantly altered climate conditions, the timing and nature of these variations are highly variable from region to region, and the notion of the Little Ice Age as a globally synchronous cold period has all but been dismissed.In China, warm-weather crops such as oranges were abandoned in Jiangxi Province, where they had been grown for centuries.[45] Also, the two periods of most frequent typhoon strikes in Guangdong coincide with two of the coldest and driest periods in northern and central China (1660–1680, 1850–1880).[46]
In Pakistan, the Balochistan province became colder and the native Baloch people started mass migration and settled along the Indus River in Sindh and Punjab provinces.[47]
Southern Hemisphere
Scientific works point out cold spells and climate changes in areas of the Southern Hemisphere and their correlation to the Little Ice Age.Africa
In Ethiopia and Mauritania, permanent snow was reported on mountain peaks at levels where it does not occur today.[45] Timbuktu, an important city on the trans-Saharan caravan route, was flooded at least 13 times by the Niger River; there are no records of similar flooding before or since.[45]In Southern Africa, sediment cores retrieved from Lake Malawi show colder conditions between 1570 and 1820, suggesting the Lake Malawi records "further support, and extend, the global expanse of the Little Ice Age."[48] A novel 3,000-year temperature reconstruction method, based on the rate of stalagmite growth in a cold cave in South Africa, further suggests a cold period from 1500 to 1800 "characterizing the South African Little Ice age."[49]
Antarctica

CO2 mixing ratios at Law Dome
The Siple Dome (SD) had a climate event with an onset time that is coincident with that of the Little Ice Age in the North Atlantic based on a correlation with the GISP2 record. The event is the most dramatic climate event in the SD Holocene glaciochemical record.[52] The Siple Dome ice core also contained its highest rate of melt layers (up to 8%) between 1550 and 1700, most likely because of warm summers.[53] Law Dome ice cores show lower levels of CO2 mixing ratios from 1550 to 1800, which Etheridge and Steele conjecture are "probably as a result of colder global climate."[54]
Sediment cores in Bransfield Basin, Antarctic Peninsula, have neoglacial indicators by diatom and sea-ice taxa variations during the Little Ice Age.[55] The MES stable isotope record suggests that the Ross Sea region experienced 1.6 ± 1.4 °C cooler average temperatures during the Little Ice Age, compared to the last 150 years to now.[56]
Australia and New Zealand
Limited evidence describes conditions in Australia. Lake records in Victoria suggest that conditions, at least in the south of the state, were wet and/or unusually cool. In the north, evidence suggests fairly dry conditions, but coral cores from the Great Barrier Reef show similar rainfall as today but with less variability. A study that analyzed isotopes in Great Barrier Reef corals suggested that increased water vapor transport from southern tropical oceans to the poles contributed to the Little Ice Age.[57] Borehole reconstructions from Australia suggest that over the last 500 years, the 17th century was the coldest on the continent, but the borehole temperature reconstruction method does not show good agreement between the Northern and Southern Hemispheres.[58]On the west coast of the Southern Alps of New Zealand, the Franz Josef glacier advanced rapidly during the Little Ice Age and reached its maximum extent in the early 18th century, in one of the few cases of a glacier thrusting into a rain forest.[30] Based on dating of a yellow-green lichen of the Rhizocarpon subgenus, the Mueller Glacier, on the eastern flank of the Southern Alps within Aoraki/Mount Cook National Park, is considered to have been at its maximum extent between 1725 and 1730.[59]
Pacific Islands
Sea-level data for the Pacific Islands suggest that sea level in the region fell, possibly in two stages, between 1270 and 1475. This was associated with a 1.5 °C fall in temperature (determined from oxygen-isotope analysis) and an observed increase in El Niño frequency.[60] Tropical Pacific coral records indicate the most frequent, intense El Niño-Southern Oscillation activity in the mid-seventeenth century.[61]South America
Tree-ring data from Patagonia show cold episodes between 1270 and 1380 and from 1520 to 1670, contemporary with the events in the Northern Hemisphere.[62][63] Eight sediment cores taken from Puyehue Lake have been interpreted as showing a humid period from 1470 to 1700, which the authors describe as a regional marker of the onset of the Little Ice Age.[64] A 2009 paper details cooler and wetter conditions in southeastern South America between 1550 and 1800, citing evidence obtained via several proxies and models.[65] 18O records from three Andean ice cores show a cool period from 1600–1800 [66]Although only anecdotal evidence, in 1675 the Spanish explorer Antonio de Vea entered San Rafael Lagoon through Río Témpanos (Spanish for "Ice Floe River") without mentioning any ice floe but stating that the San Rafael Glacier did not reach far into the lagoon. In 1766, another expedition noticed that the glacier reached the lagoon and calved into large icebergs. Hans Steffen visited the area in 1898, noticing that the glacier penetrated far into the lagoon. Such historical records indicate a general cooling in the area between 1675 and 1898: "The recognition of the LIA in northern Patagonia, through the use of documentary sources, provides important, independent evidence for the occurrence of this phenomenon in the region."[67] As of 2001, the border of the glacier had significantly retreated as compared to the borders of 1675.[67]
Possible causes
Scientists have tentatively identified these possible causes of the Little Ice Age: orbital cycles, decreased solar activity, increased volcanic activity, altered ocean current flows,[68] the inherent variability of global climate, and reforestation following decreases in the human population.Orbital cycles
Orbital forcing from cycles in the earth's orbit around the sun has, for the past 2,000 years, caused a long-term northern hemisphere cooling trend that continued through the Middle Ages and the Little Ice Age. The rate of Arctic cooling is roughly 0.02 °C per century.[69] This trend could be extrapolated to continue into the future, possibly leading to a full ice age, but the twentieth-century instrumental temperature record shows a sudden reversal of this trend, with a rise in global temperatures attributed to greenhouse gas emissions.[69]Solar activity

Solar activity events recorded in radiocarbon

The Maunder minimum in a 400-year history of sunspot numbers
There is still a very poor understanding of the correlation between low sunspot activity and cooling temperatures.[70][71] During the period 1645–1715, in the middle of the Little Ice Age, there was a period of low solar activity known as the Maunder Minimum. The Spörer Minimum has also been identified with a significant cooling period between 1460 and 1550.[72] Other indicators of low solar activity during this period are levels of the isotopes carbon-14 and beryllium-10.[73]
Volcanic activity
In a 2012 paper, Miller et al. link the Little Ice Age to an "unusual 50-year-long episode with four large sulfur-rich explosive eruptions, each with global sulfate loading >60 Tg" and notes that "large changes in solar irradiance are not required."[6]Throughout the Little Ice Age, the world experienced heightened volcanic activity.[74] When a volcano erupts, its ash reaches high into the atmosphere and can spread to cover the whole earth. The ash cloud blocks out some of the incoming solar radiation, leading to worldwide cooling that can last up to two years after an eruption. Also emitted by eruptions is sulfur, in the form of sulfur dioxide gas. When it reaches the stratosphere, it turns into sulfuric acid particles, which reflect the sun's rays, further reducing the amount of radiation reaching Earth's surface.
A recent study found that an especially massive tropical volcanic eruption in 1257, possibly of the now-extinct Mount Samalas near Mount Rinjani, both in Lombok, Indonesia, followed by three smaller eruptions in 1268, 1275, and 1284 did not allow the climate to recover. This may have caused the initial cooling, and the 1452–53 eruption of Kuwae in Vanuatu triggered a second pulse of cooling.[6][14] The cold summers can be maintained by sea-ice/ocean feedbacks long after volcanic aerosols are removed.
Other volcanoes that erupted during the era and may have contributed to the cooling include Billy Mitchell (ca. 1580), Huaynaputina (1600), Mount Parker (1641), Long Island (Papua New Guinea) (ca. 1660), and Laki (1783).[20] The 1815 eruption of Tambora, also in Indonesia, blanketed the atmosphere with ash; the following year, 1816, came to be known as the Year Without a Summer, when frost and snow were reported in June and July in both New England and Northern Europe.
Ocean circulation

Thermohaline circulation or Oceanic conveyor belt illustrated
Another possibility is that there was a slowing of thermohaline circulation.[39][68][75][76] The circulation could have been interrupted by the introduction of a large amount of fresh water into the North Atlantic, possibly caused by a period of warming before the Little Ice Age known as the Medieval Warm Period.[30][77][78] There is some concern that a shutdown of thermohaline circulation could happen again as a result of the present warming period.[79][80]
























