| T lymphocyte cell | |
|---|---|
 Scanning electron micrograph of a human T cell | |
 Scanning electron micrograph of a red blood cell (left), a platelet (center), and a T lymphocyte (right); colorized | |
| Details | |
| System | Immune system |
| Identifiers | |
| Latin | lymphocytus T |
| MeSH | D013601 |
| TH | H2.00.04.1.02007 |
| FMA | 62870 |
A T cell is a type of lymphocyte. T cells are one of the important white blood cells of the immune system and play a central role in the adaptive immune response. T cells can be distinguished from other lymphocytes by the presence of a T-cell receptor (TCR) on their cell surface.
T cells are born from hematopoietic stem cells, found in the bone marrow. Developing T cells then migrate to the thymus gland to mature. T cells derive their name from this organ where they develop (or mature). After migration to the thymus, the precursor cells mature into several distinct types of T cells. T cell differentiation also continues after they have left the thymus. Groups of specific, differentiated T cell subtypes have a variety of important functions in controlling and shaping the immune response.
One of these functions is immune-mediated cell death, and it is carried out by two major subtypes: CD8+ "killer" and CD4+ "helper" T cells. (These are named for the presence of the cell surface proteins CD8 or CD4.) CD8+ T cells, also known as "killer T cells", are cytotoxic – this means that they are able to directly kill virus-infected cells, as well as cancer cells. CD8+ T cells are also able to use small signaling proteins, known as cytokines, to recruit other types of cells when mounting an immune response. A different population of T cells, the CD4+ T cells, function as "helper cells". Unlike CD8+ killer T cells, these CD4+ helper T cells function by indirectly killing cells identified as foreign: they determine if and how other parts of the immune system respond to a specific, perceived threat. Helper T cells also use cytokine signaling to influence regulatory B cells directly, and other cell populations indirectly.
Regulatory T cells are yet another distinct population of T cells that provide the critical mechanism of tolerance, whereby immune cells are able to distinguish invading cells from "self". This prevents immune cells from inappropriately reacting against one's own cells, known as an "autoimmune" response. For this reason, these regulatory T cells have also been called "suppressor" T cells. These same regulatory T cells can also be co-opted by cancer cells to prevent the recognition of, and an immune response against, tumor cells.
Development
Origin, early development and migration to the thymus
All T cells originate from c-kit+Sca1+ haematopoietic stem cells (HSC) which reside in the bone marrow. In some cases, the origin might be the fetal liver during embryonic development. The HSC then differentiate into multipotent progenitors (MPP) which retain the potential to become both myeloid and lymphoid cells. The process of differentiation then proceeds to a common lymphoid progenitor (CLP), which can only differentiate into T, B or NK cells. These CLP cells then migrate via the blood to the thymus, where they engraft. The earliest cells which arrived in the thymus are termed double-negative, as they express neither the CD4 nor CD8 co-receptor. The newly arrived CLP cells are CD4−CD8−CD44+CD25−ckit+ cells, and are termed early thymic progenitor (ETP) cells. These cells will then undergo a round of division and downregulate c-kit and are termed DN1 cells.
TCR development
A critical step in T cell maturation is making a functional T cell receptor (TCR). Each mature T cell will ultimately contain a unique TCR that reacts to a random pattern, allowing the immune system to recognize many different types of pathogens.
The TCR consists of two major components, the alpha and beta chains. These both contain random elements designed to produce a wide variety of different TCRs, but also therefore must be tested to make sure they work at all. First, T cells attempt to create a functional beta chain, testing it against a mock alpha chain. Then they attempt to create a functional alpha chain. Once a working TCR has been produced, T cells then must show their TCR can recognize the body’s MHC complex (positive selection) and that it does not react to self proteins (negative selection).
TCR-Beta selection
At the DN2 stage (CD44+CD25+), cells upregulate the recombination genes RAG1 and RAG2 and re-arrange the TCRβ locus, combining V-D-J and constant region genes in an attempt to create a functional TCRβ chain. As the developing thymocyte progresses through to the DN3 stage (CD44−CD25+), the T cell expresses an invariant α-chain called pre-Tα alongside the TCRβ gene. If the rearranged β-chain successfully pairs with the invariant α-chain, signals are produced which cease rearrangement of the β-chain (and silences the alternate allele). Although these signals require this pre-TCR at the cell surface, they are independent of ligand binding to the pre-TCR. If the pre-TCR forms, then the cell downregulates CD25 and is termed a DN4 cell (CD25−CD44−). These cells then undergo a round of proliferation and begin to re-arrange the TCRα locus.
Positive selection
Double-positive thymocytes (CD4+/CD8+) migrate deep into the thymic cortex, where they are presented with self-antigens. These self-antigens are expressed by thymic cortical epithelial cells on MHC molecules on the surface of cortical epithelial cells. Only those thymocytes that interact with MHC-I or MHC-II will receive a vital "survival signal". All that cannot (if they do not interact strongly enough) will die by "death by neglect" (no survival signal). This process ensures that the selected T cells will have an MHC affinity that can serve useful functions in the body (i.e., the cells must be able to interact with MHC and peptide complexes to affect immune responses). The vast majority of developing thymocytes will die during this process. The process of positive selection takes a number of days.
A thymocyte's fate is determined during positive selection. Double-positive cells (CD4+/CD8+) that interact well with MHC class II molecules will eventually become CD4+ cells, whereas thymocytes that interact well with MHC class I molecules mature into CD8+ cells. A T cell becomes a CD4+ cell by down-regulating expression of its CD8 cell surface receptors. If the cell does not lose its signal, it will continue downregulating CD8 and become a CD4+, single positive cell.
This process does not remove thymocytes that may cause autoimmunity. The potentially autoimmune cells are removed by the process of negative selection, which occurs in the thymic medulla (discussed below).
Negative selection
Negative selection removes thymocytes that are capable of strongly binding with "self" MHC peptides. Thymocytes that survive positive selection migrate towards the boundary of the cortex and medulla in the thymus. While in the medulla, they are again presented with a self-antigen presented on the MHC complex of medullary thymic epithelial cells (mTECs). mTECs must be AIRE+ to properly express self-antigens from all tissues of the body on their MHC class I peptides. Some mTECs are phagocytosed by thymic dendritic cells; this allows for presentation of self-antigens on MHC class II molecules (positively selected CD4+ cells must interact with MHC class II molecules, thus APCs, which possess MHC class II, must be present for CD4+ T-cell negative selection). Thymocytes that interact too strongly with the self-antigen receive an apoptotic signal that leads to cell death. However, some of these cells are selected to become Treg cells. The remaining cells exit the thymus as mature naive T cells, also known as recent thymic emigrants. This process is an important component of central tolerance and serves to prevent the formation of self-reactive T cells that are capable of inducing autoimmune diseases in the host.
β-selection is the first checkpoint, where the T cells that are able to form a functional pre-TCR with an invariant alpha chain and a functional beta chain are allowed to continue development in the thymus. Next, positive selection checks that T cells have successfully rearranged their TCRα locus and are capable of recognizing peptide-MHC complexes with appropriate affinity. Negative selection in the medulla then obliterates T cells that bind too strongly to self-antigens expressed on MHC molecules. These selection processes allow for tolerance of self by the immune system. Typical T cells that leave the thymus (via the corticomedullary junction) are self-restricted, self-tolerant, and single positive.
Thymic output
About 98% of thymocytes die during the development processes in the thymus by failing either positive selection or negative selection, whereas the other 2% survive and leave the thymus to become mature immunocompetent T cells. The thymus contributes fewer cells as a person ages. As the thymus shrinks by about 3% a year throughout middle age, a corresponding fall in the thymic production of naive T cells occurs, leaving peripheral T cell expansion and regeneration to play a greater role in protecting older people.
Types of T cell
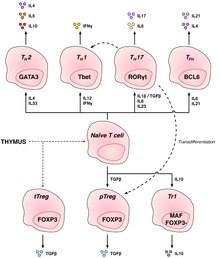
T cells are grouped into a series of subsets based on their function. CD4 and CD8 T cells are selected in the thymus, but undergo further differentiation in the periphery to specialized cells which have different functions. T cell subsets were initially defined by function, but also have associated gene or protein expression patterns.
Conventional adaptive T cells
Helper CD4+ T cells
T helper cells (TH cells) assist other lymphocytes, including maturation of B cells into plasma cells and memory B cells, and activation of cytotoxic T cells and macrophages. These cells are also known as CD4+ T cells as they express the CD4 glycoprotein on their surfaces. Helper T cells become activated when they are presented with peptide antigens by MHC class II molecules, which are expressed on the surface of antigen-presenting cells (APCs). Once activated, they divide rapidly and secrete cytokines that regulate or assist the immune response. These cells can differentiate into one of several subtypes, which have different roles. Cytokines direct T cells into particular subtypes.
| Cell type | Cytokines Produced | Key Transcription Factor | Role in immune defense | Related diseases |
|---|---|---|---|---|
| Th1 | IFNγ | Tbet | Produce an inflammatory response, key for defense against intracellular bacteria, viruses and cancer. | MS, Type 1 diabetes |
| Th2 | IL-4 | GATA-3 | Aid the differentiation and antibody production by B cells | Asthma and other allergic diseases |
| Th17 | IL-17 | RORγt | Defense against gut pathogens and at mucosal barriers | MS, Rheumatoid Arthritis, Psoriasis |
| Th9 | IL-9 | IRF4, PU.1 | Defense against helminths (parasitic worms) | Multiple Sclerosis |
| Tfh | IL-21, IL-4 | Bcl-6 | Help B cells produce antibodies | Asthma and other allergic diseases |
Cytotoxic CD8+ T cells

Cytotoxic T cells (TC cells, CTLs, T-killer cells, killer T cells) destroy virus-infected cells and tumor cells, and are also implicated in transplant rejection. These cells are defined by the expression of the CD8 protein on their cell surface. Cytotoxic T cells recognize their targets by binding to short peptides (8-11 amino acids in length) associated with MHC class I molecules, present on the surface of all nucleated cells. Cytotoxic T cells also produce the key cytokines IL-2 and IFNγ. These cytokines influence the effector functions of other cells, in particular macrophages and NK cells.
Memory T cells
Antigen-naive T cells expand and differentiate into memory and effector T cells after they encounter their cognate antigen within the context of an MHC molecule on the surface of a professional antigen presenting cell (e.g. a dendritic cell). Appropriate co-stimulation must be present at the time of antigen encounter for this process to occur. Historically, memory T cells were thought to belong to either the effector or central memory subtypes, each with their own distinguishing set of cell surface markers (see below). Subsequently, numerous new populations of memory T cells were discovered including tissue-resident memory T (Trm) cells, stem memory TSCM cells, and virtual memory T cells. The single unifying theme for all memory T cell subtypes is that they are long-lived and can quickly expand to large numbers of effector T cells upon re-exposure to their cognate antigen. By this mechanism they provide the immune system with "memory" against previously encountered pathogens. Memory T cells may be either CD4+ or CD8+ and usually express CD45RO.
Memory T cell subtypes:
- Central memory T cells (TCM cells) express CD45RO, C-C chemokine receptor type 7 (CCR7), and L-selectin (CD62L). Central memory T cells also have intermediate to high expression of CD44. This memory subpopulation is commonly found in the lymph nodes and in the peripheral circulation. (Note- CD44 expression is usually used to distinguish murine naive from memory T cells).
- Effector memory T cells (TEM cells and TEMRA cells) express CD45RO but lack expression of CCR7 and L-selectin. They also have intermediate to high expression of CD44. These memory T cells lack lymph node-homing receptors and are thus found in the peripheral circulation and tissues. TEMRA stands for terminally differentiated effector memory cells re-expressing CD45RA, which is a marker usually found on naive T cells.
- Tissue resident memory T cells (TRM) occupy tissues (skin, lung, etc.) without recirculating. One cell surface marker that has been associated with TRM is the intern αeβ7, also known as CD103.
- Virtual memory T cells differ from the other memory subsets in that they do not originate following a strong clonal expansion event. Thus, although this population as a whole is abundant within the peripheral circulation, individual virtual memory T cell clones reside at relatively low frequencies. One theory is that homeostatic proliferation gives rise to this T cell population. Although CD8 virtual memory T cells were the first to be described, it is now known that CD4 virtual memory cells also exist.
Regulatory CD4+ T cells
Regulatory T cells are crucial for the maintenance of immunological tolerance. Their major role is to shut down T cell-mediated immunity toward the end of an immune reaction and to suppress autoreactive T cells that escaped the process of negative selection in the thymus.
Two major classes of CD4+ Treg cells have been described — FOXP3+ Treg cells and FOXP3− Treg cells.
Regulatory T cells can develop either during normal development in the thymus, and are then known as thymic Treg cells, or can be induced peripherally and are called peripherally derived Treg cells. These two subsets were previously called "naturally occurring" and "adaptive" (or "induced"), respectively. Both subsets require the expression of the transcription factor FOXP3 which can be used to identify the cells. Mutations of the FOXP3 gene can prevent regulatory T cell development, causing the fatal autoimmune disease IPEX.
Several other types of T cells have suppressive activity, but do not express FOXP3 constitutively. These include Tr1 and Th3 cells, which are thought to originate during an immune response and act by producing suppressive molecules. Tr1 cells are associated with IL-10, and Th3 cells are associated with TGF-beta. Recently, Treg17 cells have been added to this list.
Innate-like T cells
Innate-like T cells or unconventional T cells represent some subsets of T cells that behave differently in immunity. They trigger rapid immune responses, regardless of the major histocompatibility complex (MHC) expression, unlike their conventional counterparts (CD4 T helper cells and CD8 cytotoxic T cells), which are dependent on the recognition of peptide antigens in the context of the MHC molecule. Overall, there are three large populations of unconventional T cells: NKT cells, MAIT cells, and gammadelta T cells. Now, their functional roles are already being well established in the context of infections and cancer. Furthermore, these T cell subsets are being translated into many therapies against malignancies such as leukemia, for example.
Natural killer T cell
Natural killer T cells (NKT cells – not to be confused with natural killer cells of the innate immune system) bridge the adaptive immune system with the innate immune system. Unlike conventional T cells that recognize protein peptide antigens presented by major histocompatibility complex (MHC) molecules, NKT cells recognize glycolipid antigens presented by CD1d. Once activated, these cells can perform functions ascribed to both helper and cytotoxic T cells: cytokine production and release of cytolytic/cell killing molecules. They are also able to recognize and eliminate some tumor cells and cells infected with herpes viruses.
Mucosal associated invariant T cells
Mucosal associated invariant T (MAIT) cells display innate, effector-like qualities. In humans, MAIT cells are found in the blood, liver, lungs, and mucosa, defending against microbial activity and infection. The MHC class I-like protein, MR1, is responsible for presenting bacterially-produced vitamin B metabolites to MAIT cells. After the presentation of foreign antigen by MR1, MAIT cells secrete pro-inflammatory cytokines and are capable of lysing bacterially-infected cells. MAIT cells can also be activated through MR1-independent signaling. In addition to possessing innate-like functions, this T cell subset supports the adaptive immune response and has a memory-like phenotype. Furthermore, MAIT cells are thought to play a role in autoimmune diseases, such as multiple sclerosis, arthritis and inflammatory bowel disease, although definitive evidence is yet to be published.
Gamma delta T cells
Gamma delta T cells (γδ T cells) represent a small subset of T cells which possess a γδ TCR rather than the αβ TCR on the cell surface. The majority of T cells express αβ TCR chains. This group of T cells is much less common in humans and mice (about 2% of total T cells) and are found mostly in the gut mucosa, within a population of intraepithelial lymphocytes. In rabbits, sheep, and chickens, the number of γδ T cells can be as high as 60% of total T cells. The antigenic molecules that activate γδ T cells are still mostly unknown. However, γδ T cells are not MHC-restricted and seem to be able to recognize whole proteins rather than requiring peptides to be presented by MHC molecules on APCs. Some murine γδ T cells recognize MHC class IB molecules. Human γδ T cells which use the Vγ9 and Vδ2 gene fragments constitute the major γδ T cell population in peripheral blood, and are unique in that they specifically and rapidly respond to a set of nonpeptidic phosphorylated isoprenoid precursors, collectively named phosphoantigens, which are produced by virtually all living cells. The most common phosphoantigens from animal and human cells (including cancer cells) are isopentenyl pyrophosphate (IPP) and its isomer dimethylallyl pyrophosphate (DMPP). Many microbes produce the highly active compound hydroxy-DMAPP (HMB-PP) and corresponding mononucleotide conjugates, in addition to IPP and DMAPP. Plant cells produce both types of phosphoantigens. Drugs activating human Vγ9/Vδ2 T cells comprise synthetic phosphoantigens and aminobisphosphonates, which upregulate endogenous IPP/DMAPP.
Activation

Activation of CD4+ T cells occurs through the simultaneous engagement of the T-cell receptor and a co-stimulatory molecule (like CD28, or ICOS) on the T cell by the major histocompatibility complex (MHCII) peptide and co-stimulatory molecules on the APC. Both are required for production of an effective immune response; in the absence of co-stimulation, T cell receptor signalling alone results in anergy. The signalling pathways downstream from co-stimulatory molecules usually engages the PI3K pathway generating PIP3 at the plasma membrane and recruiting PH domain containing signaling molecules like PDK1 that are essential for the activation of PKC-θ, and eventual IL-2 production. Optimal CD8+ T cell response relies on CD4+ signalling. CD4+ cells are useful in the initial antigenic activation of naive CD8 T cells, and sustaining memory CD8+ T cells in the aftermath of an acute infection. Therefore, activation of CD4+ T cells can be beneficial to the action of CD8+ T cells.
The first signal is provided by binding of the T cell receptor to its cognate peptide presented on MHCII on an APC. MHCII is restricted to so-called professional antigen-presenting cells, like dendritic cells, B cells, and macrophages, to name a few. The peptides presented to CD8+ T cells by MHC class I molecules are 8–13 amino acids in length; the peptides presented to CD4+ cells by MHC class II molecules are longer, usually 12–25 amino acids in length, as the ends of the binding cleft of the MHC class II molecule are open.
The second signal comes from co-stimulation, in which surface receptors on the APC are induced by a relatively small number of stimuli, usually products of pathogens, but sometimes breakdown products of cells, such as necrotic-bodies or heat shock proteins. The only co-stimulatory receptor expressed constitutively by naive T cells is CD28, so co-stimulation for these cells comes from the CD80 and CD86 proteins, which together constitute the B7 protein, (B7.1 and B7.2, respectively) on the APC. Other receptors are expressed upon activation of the T cell, such as OX40 and ICOS, but these largely depend upon CD28 for their expression. The second signal licenses the T cell to respond to an antigen. Without it, the T cell becomes anergic, and it becomes more difficult for it to activate in future. This mechanism prevents inappropriate responses to self, as self-peptides will not usually be presented with suitable co-stimulation. Once a T cell has been appropriately activated (i.e. has received signal one and signal two) it alters its cell surface expression of a variety of proteins. Markers of T cell activation include CD69, CD71 and CD25 (also a marker for Treg cells), and HLA-DR (a marker of human T cell activation). CTLA-4 expression is also up-regulated on activated T cells, which in turn outcompetes CD28 for binding to the B7 proteins. This is a checkpoint mechanism to prevent over activation of the T cell. Activated T cells also change their cell surface glycosylation profile.
The T cell receptor exists as a complex of several proteins. The actual T cell receptor is composed of two separate peptide chains, which are produced from the independent T cell receptor alpha and beta (TCRα and TCRβ) genes. The other proteins in the complex are the CD3 proteins: CD3εγ and CD3εδ heterodimers and, most important, a CD3ζ homodimer, which has a total of six ITAM motifs. The ITAM motifs on the CD3ζ can be phosphorylated by Lck and in turn recruit ZAP-70. Lck and/or ZAP-70 can also phosphorylate the tyrosines on many other molecules, not least CD28, LAT and SLP-76, which allows the aggregation of signalling complexes around these proteins.
Phosphorylated LAT recruits SLP-76 to the membrane, where it can then bring in PLC-γ, VAV1, Itk and potentially PI3K. PLC-γ cleaves PI(4,5)P2 on the inner leaflet of the membrane to create the active intermediaries diacylglycerol (DAG), inositol-1,4,5-trisphosphate (IP3); PI3K also acts on PIP2, phosphorylating it to produce phosphatidlyinositol-3,4,5-trisphosphate (PIP3). DAG binds and activates some PKCs. Most important in T cells is PKC-θ, critical for activating the transcription factors NF-κB and AP-1. IP3 is released from the membrane by PLC-γ and diffuses rapidly to activate calcium channel receptors on the ER, which induces the release of calcium into the cytosol. Low calcium in the endoplasmic reticulum causes STIM1 clustering on the ER membrane and leads to activation of cell membrane CRAC channels that allows additional calcium to flow into the cytosol from the extracellular space. This aggregated cytosolic calcium binds calmodulin, which can then activate calcineurin. Calcineurin, in turn, activates NFAT, which then translocates to the nucleus. NFAT is a transcription factor that activates the transcription of a pleiotropic set of genes, most notable, IL-2, a cytokine that promotes long-term proliferation of activated T cells.
PLC-γ can also initiate the NF-κB pathway. DAG activates PKC-θ, which then phosphorylates CARMA1, causing it to unfold and function as a scaffold. The cytosolic domains bind an adapter BCL10 via CARD (Caspase activation and recruitment domains) domains; that then binds TRAF6, which is ubiquitinated at K63. This form of ubiquitination does not lead to degradation of target proteins. Rather, it serves to recruit NEMO, IKKα and -β, and TAB1-2/ TAK1. TAK 1 phosphorylates IKK-β, which then phosphorylates IκB allowing for K48 ubiquitination: leads to proteasomal degradation. Rel A and p50 can then enter the nucleus and bind the NF-κB response element. This coupled with NFAT signaling allows for complete activation of the IL-2 gene.
While in most cases activation is dependent on TCR recognition of antigen, alternative pathways for activation have been described. For example, cytotoxic T cells have been shown to become activated when targeted by other CD8 T cells leading to tolerization of the latter.
In spring 2014, the T-Cell Activation in Space (TCAS) experiment was launched to the International Space Station on the SpaceX CRS-3 mission to study how "deficiencies in the human immune system are affected by a microgravity environment".
T cell activation is modulated by reactive oxygen species.
Antigen discrimination
A unique feature of T cells is their ability to discriminate between healthy and abnormal (e.g. infected or cancerous) cells in the body. Healthy cells typically express a large number of self derived pMHC on their cell surface and although the T cell antigen receptor can interact with at least a subset of these self pMHC, the T cell generally ignores these healthy cells. However, when these very same cells contain even minute quantities of pathogen derived pMHC, T cells are able to become activated and initiate immune responses. The ability of T cells to ignore healthy cells but respond when these same cells contain pathogen (or cancer) derived pMHC is known as antigen discrimination. The molecular mechanisms that underlie this process are controversial.
Clinical significance
Deficiency
Causes of T cell deficiency include lymphocytopenia of T cells and/or defects on function of individual T cells. Complete insufficiency of T cell function can result from hereditary conditions such as severe combined immunodeficiency (SCID), Omenn syndrome, and cartilage–hair hypoplasia. Causes of partial insufficiencies of T cell function include acquired immune deficiency syndrome (AIDS), and hereditary conditions such as DiGeorge syndrome (DGS), chromosomal breakage syndromes (CBSs), and B cell and T cell combined disorders such as ataxia-telangiectasia (AT) and Wiskott–Aldrich syndrome (WAS).
The main pathogens of concern in T cell deficiencies are intracellular pathogens, including Herpes simplex virus, Mycobacterium and Listeria. Also, fungal infections are also more common and severe in T cell deficiencies.
Cancer
Cancer of T cells is termed T-cell lymphoma, and accounts for perhaps one in ten cases of non-Hodgkin lymphoma. The main forms of T cell lymphoma are:
- Extranodal T cell lymphoma
- Cutaneous T cell lymphomas: Sézary syndrome and Mycosis fungoides
- Anaplastic large cell lymphoma
- Angioimmunoblastic T cell lymphoma
Exhaustion
T cell exhaustion is a state of dysfunctional T cells. It is characterized by progressive loss of function, changes in transcriptional profiles and sustained expression of inhibitory receptors. At first cells lose their ability to produce IL-2 and TNFα followed by the loss of high proliferative capacity and cytotoxic potential, eventually leading to their deletion. Exhausted T cells typically indicate higher levels of CD43, CD69 and inhibitory receptors combined with lower expression of CD62L and CD127. Exhaustion can develop during chronic infections, sepsis and cancer. Exhausted T cells preserve their functional exhaustion even after repeated antigen exposure.
During chronic infection and sepsis
T cell exhaustion can be triggered by several factors like persistent antigen exposure and lack of CD4 T cell help. Antigen exposure also has effect on the course of exhaustion because longer exposure time and higher viral load increases the severity of T cell exhaustion. At least 2–4 weeks exposure is needed to establish exhaustion. Another factor able to induce exhaustion are inhibitory receptors including programmed cell death protein 1 (PD1), CTLA-4, T cell membrane protein-3 (TIM3), and lymphocyte activation gene 3 protein (LAG3). Soluble molecules such as cytokines IL-10 or TGF-β are also able to trigger exhaustion. Last known factors that can play a role in T cell exhaustion are regulatory cells. Treg cells can be a source of IL-10 and TGF-β and therefore they can play a role in T cell exhaustion. Furthermore, T cell exhaustion is reverted after depletion of Treg cells and blockade of PD1. T cell exhaustion can also occur during sepsis as a result of cytokine storm. Later after the initial septic encounter anti-inflammatory cytokines and pro-apoptotic proteins take over to protect the body from damage. Sepsis also carries high antigen load and inflammation. In this stage of sepsis T cell exhaustion increases. Currently there are studies aiming to utilize inhibitory receptor blockades in treatment of sepsis.
During transplantation
While during infection T cell exhaustion can develop following persistent antigen exposure after graft transplant similar situation arises with alloantigen presence. It was shown that T cell response diminishes over time after kidney transplant. These data suggest T cell exhaustion plays an important role in tolerance of a graft mainly by depletion of alloreactive CD8 T cells. Several studies showed positive effect of chronic infection on graft acceptance and its long-term survival mediated partly by T cell exhaustion. It was also shown that recipient T cell exhaustion provides sufficient conditions for NK cell transfer. While there are data showing that induction of T cell exhaustion can be beneficial for transplantation it also carries disadvantages among which can be counted increased number of infections and the risk of tumor development.
During cancer
During cancer T cell exhaustion plays a role in tumor protection. According to research some cancer-associated cells as well as tumor cells themselves can actively induce T cell exhaustion at the site of tumor. T cell exhaustion can also play a role in cancer relapses as was shown on leukemia. Some studies have suggested that it is possible to predict relapse of leukemia based on expression of inhibitory receptors PD-1 and TIM-3 by T cells. Many experiments and clinical trials have focused on immune checkpoint blockers in cancer therapy, with some of these approved as valid therapies that are now in clinical use. Inhibitory receptors targeted by those medical procedures are vital in T cell exhaustion and blocking them can reverse these changes.















