From Wikipedia, the free encyclopedia
Silicon, 14Si
 |
| General properties |
| Pronunciation |
(SIL-ik-ən) |
| Appearance |
crystalline, reflective with bluish-tinged faces |
| Standard atomic weight (Ar, standard) |
[28.084, 28.086] conventional: 28.085 |
| Silicon in the periodic table |
|
|
| Atomic number (Z) |
14 |
| Group |
group 14 (carbon group) |
| Period |
period 3 |
| Element category |
metalloid |
| Block |
p-block |
| Electron configuration |
[Ne] 3s2 3p2 |
Electrons per shell
|
2, 8, 4 |
| Physical properties |
| Phase at STP |
solid |
| Melting point |
1687 K (1414 °C, 2577 °F) |
| Boiling point |
3538 K (3265 °C, 5909 °F) |
| Density (near r.t.) |
2.3290 g/cm3 |
| when liquid (at m.p.) |
2.57 g/cm3 |
| Heat of fusion |
50.21 kJ/mol |
| Heat of vaporization |
383 kJ/mol |
| Molar heat capacity |
19.789 J/(mol·K) |
Vapor pressure
| P (Pa) |
1 |
10 |
100 |
1 k |
10 k |
100 k |
| at T (K) |
1908 |
2102 |
2339 |
2636 |
3021 |
3537 |
|
| Atomic properties |
| Oxidation states |
4, 3, 2, 1[1] −1, −2, −3, −4 (an amphoteric oxide) |
| Electronegativity |
Pauling scale: 1.90 |
| Ionization energies |
- 1st: 786.5 kJ/mol
- 2nd: 1577.1 kJ/mol
- 3rd: 3231.6 kJ/mol
- (more)
|
| Atomic radius |
empirical: 111 pm |
| Covalent radius |
111 pm |
| Van der Waals radius |
210 pm |
|
|
| Miscellanea |
| Crystal structure |
face-centered diamond-cubic
|
| Speed of sound thin rod |
8433 m/s (at 20 °C) |
| Thermal expansion |
2.6 µm/(m·K) (at 25 °C) |
| Thermal conductivity |
149 W/(m·K) |
| Electrical resistivity |
2.3×103 Ω·m (at 20 °C)[2] |
| Band gap |
1.12 eV (at 300 K) |
| Magnetic ordering |
diamagnetic[3] |
| Magnetic susceptibility |
−3.9·10−6 cm3/mol (298 K)[4] |
| Young's modulus |
130–188 GPa[5] |
| Shear modulus |
51–80 GPa[5] |
| Bulk modulus |
97.6 GPa[5] |
| Poisson ratio |
0.064–0.28[5] |
| Mohs hardness |
6.5 |
| CAS Number |
7440-21-3 |
| History |
| Naming |
after Latin 'silex' or 'silicis', meaning flint |
| Prediction |
Antoine Lavoisier (1787) |
| Discovery and first isolation |
Jöns Jacob Berzelius[6][7] (1823) |
| Named by |
Thomas Thomson (1817) |
| Main isotopes of silicon |
|
|
|
Silicon is a
chemical element with symbol
Si and
atomic number 14. A hard and brittle crystalline solid with a blue-grey metallic lustre, it is a
tetravalent metalloid and
semiconductor. It is a member of
group 14 in the periodic table, along with
carbon above it and
germanium,
tin, and
lead
below. It is rather unreactive, though less so than germanium, and has a
very large chemical affinity for oxygen; as such, it was first prepared
and characterized in pure form only in 1823 by
Jöns Jakob Berzelius.
Its melting and boiling points of 1414 °C and 3265 °C respectively are
the second-highest among all the metalloids and nonmetals, being only
surpassed by boron (carbon sublimes rather than melts at atmospheric
pressure, albeit at a higher temperature than boron).
Silicon is the eighth most
common element in the universe by mass, but very rarely occurs as the pure element in the Earth's crust. It is most widely distributed in
dusts,
sands,
planetoids, and
planets as various forms of
silicon dioxide (silica) or
silicates. Over 90% of the Earth's crust is composed of
silicate minerals, making silicon the
second most abundant element in the Earth's crust (about 28% by mass) after
oxygen.
Most silicon is used commercially without being separated, and often
with little processing of the natural minerals. Such use includes
industrial construction with
clays,
silica sand, and
stone. Silicates are used in
Portland cement for
mortar and
stucco, and mixed with silica sand and
gravel to make
concrete for walkways, foundations, and roads. They are also used in whiteware
ceramics such as
porcelain, and in traditional
quartz-based
soda-lime glass and many other specialty
glasses. Silicon compounds such as
silicon carbide
are used as abrasives and components of high-strength ceramics. Silicon
is the basis of the widely used synthetic polymers called
silicones.
Elemental silicon also has a large impact on the modern world economy. Most free silicon is used in the
steel refining,
aluminium-casting, and fine chemical industries (often to make
fumed silica).
Even more visibly, the relatively small portion of very highly purified
elemental silicon used in semiconductor electronics (< 10%) is
essential to
integrated circuits — most computers, cell phones, and modern technology depend on it.
Silicon is an essential element in biology, although only traces are required by animals. However, various
sea sponges and microorganisms, such as
diatoms and
radiolaria, secrete skeletal structures made of silica. Silica is deposited in many plant tissues, such as in the bark and wood of
Chrysobalanaceae and the silica cells and silicified trichomes of
Cannabis sativa,
horsetails and many
grasses.
[8]
History
In 1787
Antoine Lavoisier suspected that
silica might be an oxide of a fundamental
chemical element,
[9] but the
chemical affinity of silicon for oxygen is high enough that he had no means to reduce the oxide and isolate the element.
[10] After an attempt to isolate silicon in 1808,
Sir Humphry Davy proposed the name "silicium" for silicon, from the Latin
silex,
silicis for flint, and adding the "-ium" ending because he believed it to be a metal.
[11] Most other languages use transliterated forms of Davy's name, sometimes adapted to local phonology (e.g.
German Silizium,
Turkish silisyum). A few others use instead a
calque of the Latin root (e.g.
Russian кремний, from
кремень "flint";
Greek πυριτιο from
πυρ "fire";
Finnish pii from
piikivi "flint").
[12]
In 1811,
Gay-Lussac and
Thénard are thought to have prepared impure
amorphous silicon, through the heating of recently isolated
potassium metal with
silicon tetrafluoride, but they did not purify and characterize the product, nor identify it as a new element.
[13] Silicon was given its present name in 1817 by Scottish chemist
Thomas Thomson. He retained part of Davy's name but added "-on" because he believed that silicon was a
nonmetal similar to
boron and
carbon.
[14] In 1823,
Jöns Jacob Berzelius
prepared amorphous silicon using approximately the same method as
Gay-Lussac (reducing potassium fluorosilicate with molten potassium
metal), but purifying the product to a brown powder by repeatedly
washing it.
[15] As a result, he is usually given credit for the element's discovery.
[16][17] The same year, Berzelius became the first to prepare
silicon tetrachloride;
silicon tetrafluoride had already been prepared long before in 1771 by
Carl Wilhelm Scheele by dissolving silica in
hydrofluoric acid.
[10]
Silicon in its more common crystalline form was not prepared until 31 years later, by
Deville.
[18][19] By
electrolyzing a mixture of
sodium chloride and
aluminium chloride containing approximately 10% silicon, he was able to obtain a slightly impure
allotrope of silicon in 1854.
[20] Later, more cost-effective methods have been developed to isolate several allotrope forms, the most recent being
silicene in 2010.
[21][22] Meanwhile, research on the chemistry of silicon continued;
Friedrich Wöhler discovered the first volatile hydrides of silicon, synthesising
trichlorosilane in 1857 and
silane itself in 1858, but a detailed investigation of the
silanes was only carried out in the early 20th century by
Alfred Stock, despite early speculation on the matter dating as far back as the beginnings of synthetic organic chemistry in the 1830s.
[23] Similarly, the first
organosilicon compound,
tetraethylsilane, was synthesised by
Charles Friedel and
James Crafts in 1863, but detailed characterisation of organosilicon chemistry was only done in the early 20th century by
Frederick Kipping.
[10]
Starting in the 1920s, the work of
William Lawrence Bragg on
X-ray crystallography successfully elucidated the compositions of the silicates, which had previously been known from
analytical chemistry but had not yet been understood, together with
Linus Pauling's development of
crystal chemistry and
Victor Goldschmidt's development of
geochemistry. The middle of the 20th century saw the development of the chemistry and industrial use of
siloxanes and the growing use of
silicone polymers,
elastomers, and
resins. In the late 20th century, the complexity of the crystal chemistry of
silicides was mapped, along with the solid-state chemistry of
doped semiconductors.
[10]
Because silicon is an important element in high-technology
semiconductor devices, many places in the world bear its name. For
example,
Santa Clara Valley in
California acquired the nickname
Silicon Valley
since the element is the base material used in the semiconductor
industry located there. Since then, many other locations have been
nicknamed for similar reasons.
[24]
Characteristics
Physical and atomic
Silicon crystallizes in a diamond cubic crystal structure
A silicon atom has fourteen
electrons. In the ground state, they are arranged in the electron configuration [Ne]3s
23p
2. Of these, four are
valence electrons, occupying the 3s orbital and two of the 3p orbitals. Like the other members of its group, the lighter
carbon and the heavier
germanium,
tin, and
lead, it has the same number of valence electrons as valence orbitals: hence, it can complete its
octet and obtain the stable
noble gas configuration of
argon by forming
sp3 hybrid orbitals, forming tetrahedral SiX
4 derivatives where the central silicon atom shares an electron pair with each of the four atoms it is bonded to.
[25] The first four
ionisation energies
of silicon are 786.3, 1576.5, 3228.3, and 4354.4 kJ/mol respectively;
these figures are high enough to preclude the possibility of simple
cationic chemistry for the element. Following
periodic trends,
its single-bond covalent radius of 117.6 pm is intermediate between
those of carbon (77.2 pm) and germanium (122.3 pm). The hexacoordinate
ionic radius of silicon may be considered to be 40 pm, although this
must be taken as a purely notional figure given the lack of a simple Si
4+ cation in reality.
[26]
At standard temperature and pressure, silicon is a shiny
semiconductor
with a bluish-grey metallic lustre; as typical for semiconductors, its
resistivity drops as temperature rises. This arises because silicon has a
small energy gap between its highest occupied energy levels (the
valence band) and the lowest unoccupied ones (the conduction band). The
Fermi level
is about halfway between the valence and conduction bands and is the
energy at which a state is as likely to be occupied by an electron as
not. Hence pure silicon is an insulator at room temperature. However,
doping silicon with a
pnictogen such as
phosphorus,
arsenic, or
antimony
introduces one extra electron per dopant and these may then be excited
into the conduction band either thermally or photolytically, creating an
n-type semiconductor. Similarly, doping silicon with a
group 13 element such as
boron,
aluminium, and
gallium
results in the introduction of acceptor levels that trap electrons that
may be excited from the filled valence band, creating a
p-type semiconductor. Joining n-type silicon to p-type silicon creates a
p-n junction
with a common Fermi level; electrons flow from n to p, while holes flow
from p to n, creating a voltage drop. This p-n junction thus acts as a
diode that can rectify alternating current that allows current to pass more easily one way than the other. A
transistor
is an n-p-n junction, with a thin layer of weakly p-type silicon
between two n-type regions. Biasing the emitter through a small forward
voltage and the collector through a large reverse voltage allows the
transistor to act as a
triode amplifier.
[27]
Silicon crystallises in a giant covalent structure at standard conditions, specifically in a
diamond cubic
lattice. It thus has a high melting point of 1414 °C, as a lot of
energy is required to break the strong covalent bonds and melt the
solid. It is not known to have any allotropes at standard pressure, but
several other crystal structures are known at higher pressures. The
general trend is one of increasing
coordination number with pressure, culminating in a
hexagonal close-packed allotrope at about 40
gigapascals
known as Si–VII (the standard modification being Si–I). Silicon boils
at 3265 °C: this, while high, is still lower than the temperature at
which its lighter congener
carbon sublimes (3642 °C) and silicon similarly has a lower
heat of vaporisation than carbon, consistent with the fact that the Si–Si bond is weaker than the C–C bond.
[27]
Isotopes
Naturally occurring silicon is composed of three stable
isotopes,
28Si (92.23%),
29Si (4.67%), and
30Si (3.10%).
[28] Out of these, only
29Si is of use in
NMR and
EPR spectroscopy,
[29] as it is the only one with a nuclear spin (
I =
1/2).
[30] All three are produced in stars through the
oxygen-burning process, with
28Si being made as part of the
alpha process and hence the most abundant. The fusion of
28Si with alpha particles by
photodisintegration rearrangement in stars is known as the
silicon-burning process; it is the last stage of
stellar nucleosynthesis before the rapid collapse and violent explosion of the star in question in a
type II supernova.
[31]
Twenty
radioisotopes have been characterized, with the two stablest being
32Si with a
half-life of about 150 years, and
31Si with a half-life of 2.62 hours.
[28] All of the remaining
radioactive
isotopes have half-lives that are less than seven seconds, and the
majority of these have half-lives that are less than one tenth of a
second.
[28] Silicon does not have any known
nuclear isomers.
[28] 32Si undergoes low-energy
beta decay to
32P and then stable
32S.
31Si may be produced by the
neutron activation
of natural silicon and is thus useful for quantitative analysis; it can
be easily detected by its characteristic beta decay to stable
31P, in which the emitted electron carries up to 1.48
MeV of energy.
[30]
The known isotopes of silicon range in
mass number from 22 to 44.
[28] The most common
decay mode of the isotopes with mass numbers lower than the three stable isotopes is
inverse beta decay, primarily forming aluminium isotopes (13 protons) as
decay products.
[28]
The most common decay mode for the heavier unstable isotopes is beta
decay, primarily forming phosphorus isotopes (15 protons) as decay
products.
[28]
Chemistry and compounds
Crystalline bulk silicon is rather inert, but becomes more reactive
at high temperatures. Like its neighbour aluminium, silicon forms a
thin, continuous surface layer of
silicon dioxide (SiO
2)
that protects the metal from oxidation. Thus silicon does not
measurably react with the air below 900 °C, but formation of the
vitreous dioxide rapidly increases between 950 °C and 1160 °C and when 1400 °C is reached, atmospheric
nitrogen also reacts to give the nitrides SiN and Si
3N
4. Silicon reacts with gaseous
sulfur at 600 °C and gaseous
phosphorus at 1000 °C. This oxide layer nevertheless does not prevent reaction with the
halogens;
fluorine attacks silicon vigorously at room temperature,
chlorine does so at about 300 °C, and
bromine and
iodine at about 500 °C. Silicon does not react with most aqueous acids, but is oxidised and fluorinated by a mixture of concentrated
nitric acid and
hydrofluoric acid; it readily dissolves in hot aqueous alkali to form
silicates. At high temperatures, silicon also reacts with
alkyl halides; this reaction can be catalysed by
copper to directly synthesise
organosilicon chlorides as precursors to
silicone polymers. Upon melting, silicon becomes extremely reactive, alloying with most metals to form
silicides, and reducing most metal oxides because the
heat of formation of silicon dioxide is so large. As a result, containers for liquid silicon must be made of
refractory, unreactive materials such as
zirconium dioxide or group 4, 5, and 6 borides.
[27]
Tetrahedral coordination is a major structural motif in silicon
chemistry just as it is for carbon chemistry. However, the 3p subshell
is rather more diffuse than the 2p subshell and does not hybridise as
well with the 3s subshell. As a result, the chemistry of silicon and its
heavier congeners shows significant differences from that of carbon,
[32] and thus octahedral coordination is also significant.
[27] For example, the
electronegativity
of silicon (1.90) is much less than that of carbon (2.55), because the
valence electrons of silicon are further from the nucleus than those of
carbon and hence experience smaller electrostatic forces of attraction
from the nucleus. The poor overlap of 3p orbitals also results in a much
lower tendency towards
catenation
(formation of Si–Si bonds) for silicon than for carbon due to the
concomitant weakening of the Si–Si bond compared to the C–C bond:
[33] the average Si–Si bond energy is approximately 226 kJ/mol, compared to a value of 356 kJ/mol for the C–C bond.
[34]
This results in multiply bonded silicon compounds generally being much
less stable than their carbon counterparts, an example of the
double bond rule. On the other hand, the presence of 3d orbitals in the valence shell of silicon suggests the possibility of
hypervalence, as seen in five- and six-coordinate derivatives of silicon such as
SiX−
5 and
SiF2−
6.
[33] Lastly, because of the increasing energy gap between the valence s and p
orbitals as the group is descended, the divalent state grows in
importance from carbon to lead, so that a few unstable divalent
compounds are known for silicon; this lowering of the main oxidation
state, in tandem with increasing atomic radii, results in an increase of
metallic character down the group. Silicon already shows some incipient
metallic behaviour, particularly in the behaviour of its oxide
compounds and its reaction with acids as well as bases (though this
takes some effort), and is hence often referred to as a
metalloid rather than a nonmetal.
[33]
However, metallicity does not become clear in group 14 until germanium
and dominant until tin, with the growing importance of the lower +2
oxidation state.
[10]
Silicon shows clear differences with carbon. For example,
organic chemistry has very few analogies with silicon chemistry, while
silicate minerals have a structural complexity unseen in
oxocarbons.
[10] Silicon tends to resemble germanium far more than it does carbon, and this resemblance is enhanced by the
d-block contraction resulting in the size of the germanium atom being much closer to that of the silicon atom than periodic trends would predict.
[26]
Nevertheless, there are still some differences because of the growing
importance of the divalent state in germanium compared to silicon, that
result in germanium being significantly more metallic than silicon.
Additionally, the lower Ge–O bond strength compared to the Si–O bond
strength results in the absence of "germanone" polymers that would be
analogous to
silicone polymers.
[34]
Silicides
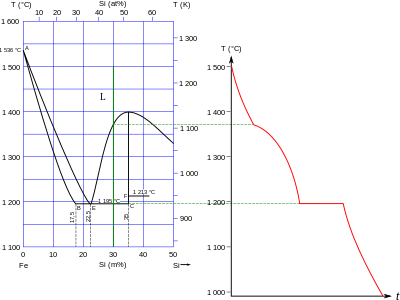
Phase diagram of the Fe–Si system
Many metal
silicides are known, most of which have formulae that cannot be explained through simple appeals to
valence: their bonding ranges from
metallic to
ionic and
covalent. Some known stoichiometries are M
6Si, M
5Si, M
4Si, M
15Si
4, M
3Si, M
5Si
2, M
2Si, M
5Si
3, M
3Si
2, MSi, M
2Si
3, MSi
2, MSi
3, and MSi
6. They are structurally more similar to the
borides than the
carbides, in keeping with the
diagonal relationship between
boron
and silicon, although the larger size of silicon than boron means that
exact structural analogies are few and far between. The heats of
formation of the silicides are usually similar to those of the borides
and carbides of the same elements, but they usually melt at lower
temperatures.
[35] Silicides are known for all stable elements in groups 1–10, with the exception of
beryllium: in particular,
uranium and the transition metals of groups 4–10 show the widest range of stoichiometries. Except for
copper, the metals in groups 11–15 do not form silicides. Most instead form
eutectic mixtures, although the heaviest
post-transition metals mercury,
thallium,
lead, and
bismuth are completely immiscible with liquid silicon.
[35]
Silicides are usually prepared by direct reaction of the elements. For example, the
alkali metals and
alkaline earth metals
react with silicon or silicon oxide to give silicides. Nevertheless,
even with these highly electropositive elements true silicon anions are
not obtainable, and most of these compounds are semiconductors. For
example, the alkali metal silicides
(M+)
4(E4−
4) contain pyramidal tricoordinate silicon in the
Si4−
4 anion, isoeelctronic with
white phosphorus, P
4.
[35][36] Metal-rich silicides tend to have isolated silicon atoms (e.g. Cu
5Si); with increasing silicon content, catenation increases, resulting in isolated clusters of two (e.g. U
3Si
2) or four silicon atoms (e.g. [K
+]
4[Si
4]
4−) at first, followed by chains (e.g. CaSi), layers (e.g. CaSi
2), or three-dimensional networks of silicon atoms spanning space (e.g. α-ThSi
2) as the silicon content rises even higher.
[35]
The silicides of the group 1 and 2 metals are usually more reactive
than the transition metal silicides. The latter usually do not react
with aqueous reagents, except for
hydrofluoric acid; however, they do react with much more aggressive reagents like liquid
potassium hydroxide, or gaseous fluorine or
chlorine
when red-hot. The pre-transition metal silicides instead readily react
with water and aqueous acids, usually producing hydrogen or silanes:
[35]
- Na2Si + 3 H2O → Na2SiO3 + 3 H2
- Mg2Si + 2 H2SO4 → 2 MgSO4 + SiH4
Products often vary with the stoichiometry of the silicide reactant. For example, Ca
2Si is polar and non-conducting and has the anti-PbCl
2 structure with single isolated silicon atoms, and reacts with water to produce
calcium hydroxide,
hydrated silicon dioxide, and hydrogen gas. CaSi with its zigzag chains
of silicon atoms instead reacts to give silanes and polymeric SiH
2, while CaSi
2
with its puckered layers of silicon atoms does not react with water,
but will react with dilute hydrochloric acid: the product is a yellow
polymeric solid with stoichiometry Si
2H
2O.
[35]
Silanes
Speculation on silicon hydride chemistry started from the 1830s, contemporary with the development of synthetic
organic chemistry.
Silane itself, as well as
trichlorosilane, were first synthesised by
Friedrich Wöhler and
Heinrich Buff in 1857 by reacting aluminium–silicon alloys with
hydrochloric acid, and characterised as SiH
4 and SiHCl
3 by
Charles Friedel and
Albert Ladenburg in 1867.
Disilane (Si
2H
6) followed in 1902, when it was first made by
Henri Moissan and
Samuel Smiles by the
protonolysis of
magnesium silicides.
Further investigation had to wait until 1916 because of the great
reactivity and thermal instability of the silanes; it was then that
Alfred Stock
began the study of silicon hydrides in earnest with new greaseless
vacuum techniques, as they were found as contaminants of his focus, the
boron hydrides. The names
silanes and
boranes are due to him, based on analogy with the
alkanes.
[23][37][38]
Moissan and Smiles' method of preparation of silanes and silane
derivatives via protonolysis of metal silicides is still used, although
the yield is lowered by the hydrolysis of the products that occurs
simultaneously, and thus the preferred route today is to treat
substituted silanes with hydride reducing agents such as
lithium aluminium hydride
in etheric solutions at low temperatures. Direct reaction of HX or RX
with silicon, possibly with a catalyst such as copper, is also a viable
method to produce substituted silanes.
[23]
The silanes comprise a
homologous series of silicon hydrides with the general formula Si
nH
2n + 2. They are all strong
reducing agents. Unbranched and branched trains are known up to
n = 8, and the cycles Si
5H
10 and Si
6H
12
are also known. The first two, silane and disilane, are colourless
gases; the heavier members of the series are volatile liquids. All
silanes are very reactive and catch fire or explode spontaneously in
air. They become less thermally stable with room temperature, so that
only silane is indefinitely stable at room temperature, although
disilane does not decompose very quickly (only 2.5% of a sample
decomposes after eight months have passed).
[23] They decompose to form polymeric
polysilicon hydride and hydrogen gas.
[39][40]
As expected from the difference in atomic weight, the silanes are less
volatile than the corresponding alkanes and boranes, but more volatile
than the corresponding germanes. They are much more reactive than the
corresponding alkanes, because the larger radius of silicon compared to
carbon facilitates
nucleophilic attack
at the silicon, the greater polarity of the Si–H bond compared to the
C–H bond, and the ability of silicon to expand its octet and hence form
adducts and lower the reaction's
activation energy.
[23]
Silane
pyrolysis
gives polymeric species and finally elemental silicon and hydrogen;
indeed ultrapure silicon is commercially produced by the pyrolysis of
silane. While the thermal decomposition of alkanes starts by the
breaking of a C–H or C–C bond and the formation of radical
intermediates, polysilanes decompose by eliminating
silenes :SiH
2
or :SiHR, as the activation energy of this process (~210 kJ/mol) is
much less than the Si–Si and Si–H bond energies. While pure silanes do
not react with pure water or dilute acids, traces of alkali catalyse
immediate hydrolysis to hydrated silicon dioxide. If the reaction is
carried out in
methanol, controlled solvolysis results in the products SiH
2(OMe)
2, SiH(OMe)
3, and Si(OMe)
4. The Si–H bond also adds to
alkenes,
a reaction which proceeds slowly and speeds up with increasing
substitution of the silane involved. At 450 °C, silane participates in
an
addition reaction with
acetone, as well as a
ring-opening reaction with
ethylene oxide.
Direct reaction of the silanes with chlorine or bromine results in
explosions at room temperature, but the reaction of silane with bromine
at −80 °C is controlled and yields bromosilane and dibromosilane. The
monohalosilanes may be formed by reacting silane with the appropriate
hydrogen halide with an Al
2X
6 catalyst, or by reacting silane with a solid
silver halide in a heated flow reactor:
[23]
- SiH4 + 2 AgCl 260 °C→ SiH3Cl + HCl + 2 Ag
Among the derivatives of silane,
iodosilane (SiH
3I) and
potassium silanide (KSiH
3)
are very useful synthetic intermediates in the production of more
complicated silicon-containing compounds: the latter is a colourless
crystalline ionic solid containing K
+ cations and
SiH−
3 anions in the
NaCl structure, and is made by the reduction of silane by
potassium metal.
[41] Additionally, the reactive hypervalent species
SiH−
5 is also known.
[23]
With suitable organic substituents it is possible to produce stable
polysilanes: they have surprisingly high electric conductivities,
arising from
sigma delocalisation of the electrons in the chain.
[42]
Halides
Silicon and
silicon carbide readily react with all four stable halogens, forming the colourless, reactive and volatile silicon tetrahalides.
[43] Silicon tetrafluoride may also be made by fluorinating the other silicon halides, and is produced by the attack of
hydrofluoric acid on glass.
[44]
Heating two different tetrahalides together also produce a random
mixture of mixed halides, which may also be produced by halogen exchange
reactions. The melting and boiling points of these species usually rise
with increasing atomic weight, though there are many exceptions: for
example, the melting and boiling points drop as one passes from SiFBr
3 through SiFClBr
2 to SiFCl
2Br.
The shift from the hypoelectronic elements in group 13 and earlier to
the group 14 elements is illustrated by the change from an infinite
ionic structure in
aluminium fluoride
to a lattice of simple covalent silicon tetrafluoride molecules, as
dictated by the lower electronegativity of aluminium than silicon, the
stoichiometry (the +4 oxidation state being too high for true ionicity),
and the smaller size of the silicon atom compared to the aluminium
atom.
[43] Silicon tetrachloride is manufactured on a huge scale as a precursor to the production of pure silicon, silicon dioxide, and some silicon
esters.
[43]
The silicon tetrahalides hydrolyse readily in water, unlike the carbon
tetrahalides, again because of the larger size of the silicon atom
rendering it more open to nucleophilic attack and the ability of the
silicon atom to expand its octet which carbon lacks.
[44] The reaction of silicon fluoride with excess
hydrofluoric acid produces the octahedral
hexafluorosilicate anion
SiF2−
6.
[44]
Analogous to the silanes, halopolysilanes Si
nX
2n + 2
are also known. While catenation in carbon compounds is maximised in
the hydrogen compounds rather than the halides, the opposite is true for
silicon, so that the halopolysilanes are known up to at least Si
14F
30, Si
6Cl
14, and Si
4Br
10.
A suggested explanation for this phenomenon is the compensation for the
electron loss of silicon to the more electronegative halogen atoms by
pi backbonding from the filled p
π orbitals on the halogen atoms to the empty d
π orbitals on silicon: this is similar to the situation of
carbon monoxide in
metal carbonyl complexes and explains their stability. These halopolysilanes may be produced by
comproportionation of silicon tetrahalides with elemental silicon, or by condensation of lighter halopolysilanes (
trimethylammonium being a useful catalyst for this reaction).
[43]
Silica
Silicon dioxide (SiO
2), also known as silica, is one of the most well-studied compounds, second only to
water. Twelve different crystal modifications of silica are known, the most common being α-
quartz, a major constituent of many rocks such as
granite and
sandstone. It is also known to occur pure as
rock crystal; impure forms are known as
rose quartz,
smoky quartz,
morion,
amethyst, and
citrine. Some poorly crystalline forms of quartz are also known, such as
chalcedony,
chrysoprase,
carnelian,
agate,
onyx,
jasper,
heliotrope, and
flint. Other modifications of silicon dioxide are known in some other minerals such as
tridymite and
cristobalite, as well as the much less common
coesite and
stishovite. Biologically generated forms are also known as
kieselguhr and
diatomaceous earth.
Vitreous silicon dioxide is known as
tektites, and
obsidian, and rarely as
lechatelierite. Some synthetic forms are known as
keatite and
W-silica.
Opals are composed of complicated crystalline aggregates of partially hydrated silicon dioxide.
[45]
Most crystalline forms of silica are made of infinite arrangements of {SiO
4}
tetrahedra (with Si at the centre) connected at their corners, with
each oxygen atom linked to two silicon atoms. In the thermodynamically
stable room-temperature form, α-quartz, these tetrahedra are linked in
intertwined helical chains with two different Si–O distances (159.7 and
161.7 pm) with a Si–O–Si angle of 144°. These helices can be either
left- or right-handed, so that individual α-quartz crystals are
optically active. At 537 °C, this transforms quickly and reversibly into
the similar β-quartz, with a change of the Si–O–Si angle to 155° but a
retention of handedness. Further heating to 867 °C results in another
reversible phase transition to β-tridymite, in which some Si–O bonds are
broken to allow for the arrangement of the {SiO
4} tetrahedra
into a more open and less dense hexagonal structure. This transition is
slow and hence tridymite occurs as a metastable mineral even below this
transition temperature; when cooled to about 120 °C it quickly and
reversibly transforms by slight displacements of individual silicon and
oxygen atoms to α-tridymite, similarly to the transition from α-quartz
to β-quartz. β-tridymite slowly transforms to cubic β-cristobalite at
about 1470 °C, which once again exists metastably below this transition
temperature and transforms at 200–280 °C to α-cristobalite via small
atomic displacements. β-cristobalite melts at 1713 °C; the freezing of
silica from the melt is quite slow and
vitrification, or the formation of a
glass, is likely to occur instead. In vitreous silica, the {SiO
4}
tetrahedra remain corner-connected, but the symmetry and periodicity of
the crystalline forms are lost. Because of the slow conversions between
these three forms, it is possible upon rapid heating to melt β-quartz
(1550 °C) or β-tridymite (1703 °C). Silica boils at approximately
2800 °C. Other high-pressure forms of silica are known, such as coesite
and stishovite: these are known in nature, formed under the shock
pressure of a meteorite impact and then rapidly quenched to preserve the
crystal structure. Similar melting and cooling of silica occurs
following
lightning strikes, forming glassy
lechatelierite. W-silica is an unstable low-density form involving {SiO
4} tetrahedra sharing opposite edges instead of corners, forming parallel chains similarly to
silicon disulfide (SiS
2) and
silicon diselenide (SiSe
2): it quickly returns to forming amorphous silica with heat or traces of water.
[45]
Condensed polysilicic acid
Silica is rather inert chemically. It is not attacked by any acids
other than hydrofluoric acid. However, it slowly dissolves in hot
concentrated alkalis, and does so rather quickly in fused metal
hydroxides or carbonates to give metal silicates. Among the elements, it
is attacked only by fluorine at room temperature to form silicon
tetrafluoride: hydrogen and carbon also react, but require temperatures
over 1000 °C to do so. Silica nevertheless reacts with many metal and
metalloid
oxides to form a wide variety of compounds important in the glass and
ceramic industries above all, but also have many other uses: for
example,
sodium silicate is often used in detergents due to its
buffering,
saponifying, and
emulsifying properties.
[45]
Silicic acids
Adding water to silica drops its melting point by around 800 °C due
to the breaking of the structure by replacing Si–O–Si linkages with
terminating Si–OH groups. Increasing water concentration results in the
formation of hydrated
silica gels and
colloidal silica dispersions. Many hydrates and
silicic acids
exist in the most dilute of aqueous solutions, but these are rather
insoluble and quickly precipitate and condense and cross-link to form
various polysilicic acids of variable combinations following the formula
[SiO
x(OH)
4−2x]
n, similar to the behaviour of
boron,
aluminium, and
iron, among other elements. Hence, although some simple silicic acids have been identified in dilute solutions, such as
orthosilicic acid Si(OH)
4 and
metasilicic acid SiO(OH)
2, none of these are likely to exist in the solid state.
[45]
Silicate minerals
About 95% of the Earth's
crustal rocks are made of silica or silicate and
aluminosilicate minerals, as reflected in oxygen, silicon, and aluminium being the three most common elements in the crust (in that order).
[46] Measured by mass, silicon makes up 27.7% of the
Earth's crust.
[47]
Pure silicon crystals are very rarely found in nature, but notable
exceptions are crystals as large as to 0.3 mm across found during
sampling gases from the
Kudriavy volcano on the island of
Iturup, one of the
Kuril Islands.
[48][49]
Silicate and aluminosilicate minerals have many different structures
and varying stoichiometry, but they may be classified following some
general principles. Tetrahedral {SiO
4} units are common to
almost all these compounds, either as discrete structures, or combined
into larger units by the sharing of corner oxygen atoms. These may be
divided into
neso-silicates (discrete {SiO
4} units) sharing no oxygen atoms,
soro-silicates (discrete {Si
2O
7} units) sharing one,
cyclo-silicates (closed ring structures) and
ino-silicates (continuous chain or ribbon structures) both sharing two,
phyllo-silicates (continuous sheets) sharing three, and
tecto-silicates
(continuous three-dimensional frameworks) sharing four. The lattice of
oxygen atoms that results is usually close-packed or close to it, with
the charge being balanced by other cations in various different
polyhedral sites according to size.
[46]
The
orthosilicates M
II
2SiO
4 (M = Be, Mg, Mn, Fe, Zn) and ZrSiO
4 are
neso-silicates. Be
2SiO
4 (
phenacite) is rather unusual as both Be
II and Si
IV
occupy tetrahedral four-coordinated sites; the other divalent cations
instead occupy six-coordinated octahedral sites and often isomorphously
replace each other as in
olivine, (Mg,Fe,Mn)
2SiO
4.
Zircon, ZrSiO
4, demands eight-coordination of the Zr
IV cations due to stoichiometry and because of their larger ionic radius (84 pm). Also significant are the
garnets, [M
II
3M
III
2(SiO
4)
3],
in which the divalent cations (e.g. Ca, Mg, Fe) are eight-coordinated
and the trivalent ones are six-coordinated (e.g. Al, Cr, Fe). Regular
coordination is not always present: for example, it is not found in Ca
2SiO
4, which mixes six- and eight-coordinate sites for Ca
II.
Soro-silicates, involving discrete double or triple tetrahedral units, are quite rare: metasilicates involving cyclic "[(SiO
3)
n]
2n−" units of corner-abutting tetrahedra forming a polygonal ring are also known.
[46]
Chain metasilicates, {SiO
2−
3}
∞, form by the corner-sharing of an indefinite chain of linked {SiO
4}
tetrahedra. Many differences arise due to the differing repeat
distances of conformation across the line of tetrahedra. A repeat
distance of two is most common, as in most
pyroxene
minerals, but repeat distances of one, three, four, five, six, seven,
nine, and twelve are also known. These chains can then link across each
other to form double chains and ribbons, as in the
asbestos minerals, involving repeated chains of cyclic tetrahedron rings.
[46]
A typical zeolite structure
Layer silicates, such as the clay minerals and the
micas,
are very common, and are often formed by horizontal cross-linking of
metasilicate chains or planar condensation of smaller units. An example
is
kaolinite [Al
2(OH)
4Si
2O
5]; in many of these minerals cation and anion replacement is common, so that for example tetrahedral Si
IV may be replaced by Al
III, octahedral Al
III by Mg
II, and OH
− by F
−. Three-dimensional framework aluminosilicates are structurally very complex; they may be conceived of as starting from the SiO
2 structure, but having replaced up to one-half of the Si
IV atoms with Al
III they require more cations to be included in the structure to balance charge. Examples include
feldspars (the most abundant minerals on the Earth),
zeolites, and
ultramarines. Many feldspars can be thought of as forming part of the ternary system NaAlSi
3O
8–KAlSi
3O
8–CaAl
2Si
2O
8. Their lattice is destroyed by high pressure prompting Al
III to undergo six-coordination rather than four-coordination, and this reaction destroying feldspars may be a reason for the
Mohorovičić discontinuity,
which would imply that the crust and mantle have the same chemical
composition but different lattices, although this is not a universally
held view. Zeolites have many polyhedral cavities in their frameworks (
truncated cuboctahedra
being most common, but other polyhedra are also known as zeolite
cavities), allowing them to include loosely bound molecules such as
water in their structure. Ultramarines alternate silicon and aluminium
atoms and include a variety of other anions such as Cl
−,
SO2−
4, and
S2−
2, but are otherwise similar to the feldspars.
[46]
Other inorganic compounds
Silicon disulfide (SiS
2)
is formed by burning silicon in gaseous sulfur at 100 °C; sublimation
of the resulting compound in nitrogen results in white, flexible long
fibres reminiscent of
asbestos
with a structure similar to W-silica. This melts at 1090 °C and
sublimes at 1250 °C; at high temperature and pressure this transforms to
a crystal structure analogous to cristobalite. However, SiS
2 lacks the variety of structures of SiO
2, and quickly hydrolyses to silica and
hydrogen sulfide. It is also ammonoloysed quickly and completely by liquid
ammonia as follows to form an
imide:
[50]
- SiS2 + 4 NH3 → Si(NH)2 + 2 NH4SH
It reacts with the sulfides of sodium, magnesium, aluminium, and iron to form metal
thiosilicates: reaction with
ethanol results in
ethylsilicate Si(OEt)
4
and hydrogen sulfide. Ethylsilicate is useful as its controlled
hydrolysis produces adhesive or film-like forms of silica. Reacting
hydrogen sulfide with silicon tetrahalides yields silicon thiohalides
such as S(SiCl)
3, cyclic Cl
2Si(μ-S)
2SiCl
2, and crystalline (SiSCl
2)
4. Despite the
double bond rule, stable organosilanethiones RR'Si=S have been made thanks to the stabilising mechanism of intermolecular coordination via an
amine group.
[50]
Silicon nitride, Si
3N
4,
can be formed by directly reacting silicon with nitrogen above 1300 °C,
but a more economical means of production is by heating silica and coke
in a stream of nitrogen and hydrogen gas at 1500 °C. It would make a
promising
ceramic
if not for the difficulty of working with and sintering it: it is
chemically near-totally inert, and even above 1000 °C it keeps its
strength, shape, and continues to be resistant to wear and corrosion. It
is very hard (9 on the
Mohs hardness scale), dissociates only at 1900 °C at 1 atm, and is quite dense (density 3.185 g/cm
3), because of its compact structure similar to that of phenacite (Be
2SiO
4). A similar refractory material is Si
2N
2O,
formed by heating silicon and silica at 1450 °C in an argon stream
containing 5% nitrogen gas, involving 4-coordinate silicon and
3-coordinate nitrogen alternating in puckered hexagonal tilings
interlinked by non-linear Si–O–Si linkages to each other.
[50]
Reacting silyl halides with ammonia or alkylammonia derivatives in
the gaseous phase or in ethanolic solution produces various volatile
silylamides, which are silicon analogues of the
amines:
[50]
- 3 SiH3Cl + 4 NH3 → N(SiH3)3 + 3 NH4Cl
- SiH3Br + 2 Me2NH → SiH3NMe2 + Me2NH2Br
- 4 SiH3I + 5 N2H4 → (SiH3)2NN(SiH3)2 + 4 N2H5I
Many such compounds have been prepared, the only known restriction
being that the nitrogen is always tertiary, and species containing the
SiH–NH group are unstable at room temperature. The stoichiometry around
the nitrogen atom in compounds such as N(SiH
3)
3is planar, which has been attributed to a p
π–d
π interaction between a lone pair on nitrogen and an empty d
π orbital on silicon. Similarly, trisilylamines are weaker as ligands than their carbon analogues, the tertiary
amines, although substitution of some SiH
3 groups by CH
3 groups mitigates this weakness. Thus, for example, N(SiH
3)
3 does not form an
adduct with
BH3 at all, while MeN(SiH
3)
2 and Me
2NSiH
3 form adducts at low temperatures that decompose upon warming. Some silicon analogues of
imines, with a Si=N double bond, are known: the first found was Bu
t2Si=N–SiBu
t3, which was discovered in 1986.
[50]
Silicon carbide (SiC) was first made by
Edward Goodrich Acheson in 1891, who named it carborundum to reference its intermediate hardness and abrasive power between
diamond (an allotrope of carbon) and
corundum (
aluminium oxide). He soon founded a company to manufacture it, and today about one million tonnes are produced each year.
[51] Silicon carbide exists in about 250 crystalline forms.
[52]
The polymorphism of SiC is characterized by a large family of similar
crystalline structures called polytypes. They are variations of the same
chemical compound that are identical in two dimensions and differ in
the third. Thus, they can be viewed as layers stacked in a certain
sequence.
[53] It is made industrially by reduction of quartz sand with excess coke or anthracite at 2000–2500 °C in an electric furnace:
[51]
- SiO2 + 2 C → Si + 2 CO
- Si + C → SiC
It is the most thermally stable binary silicon compound, only
decomposing through loss of silicon starting from around 2700 °C. It is
resistant to most aqueous acids,
phosphoric acid being an exception. It forms a protective layer of
silicon dioxide
on the surface and hence only oxidises appreciably in air above
1000 °C; removal of this layer by molten hydroxides or carbonates leads
to quick oxidation. Silicon carbide is rapidly attacked by chlorine gas,
which forms SiCl
4 and carbon at 100 °C and SiCl
4 and
CCl4
at 1000 °C. It is mostly used as an abrasive and a refractory materia,
as it is chemically stable and very strong, and it fractures to form a
very sharp cutting edge. It is also useful as an intrinsic
semiconductor, as well as an extrinsic semiconductor upon being doped.
[51] In its diamond-like behaviour it serves as an illustration of the chemical similarity between carbon and silicon.
[54]
Organosilicon compounds

A hydrosilylation reaction, in which Si–H is added to an unsaturated substrate
Because the Si–C bond is close in strength to the C–C bond,
organosilicon compounds tend to be markedly thermally and chemically
stable. For example,
tetraphenylsilane (SiPh
4) may be distilled in air even at its boiling point of 428 °C, and so can its substituted derivatives Ph
3SiCl and Ph
2SiCl
2,
which boil at 378 °C and 305 °C respectively. Furthermore, since carbon
and silicon are chemical congeners, organosilicon chemistry shows some
significant similarities with carbon chemistry, for example in the
propensity of such compounds for catenation and forming multiple bonds.
[54] However, significant differences also arise: since silicon is more
electropositive than carbon, bonds to more electronegative elements are
generally stronger with silicon than with carbon, and vice versa. Thus
the Si–F bond is significantly stronger than even the
C–F bond
and is one of the strongest single bonds, while the Si–H bond is much
weaker than the C–H bond and is readily broken. Furthermore, the ability
of silicon to expand its octet is not shared by carbon, and hence some
organosilicon reactions have no organic analogues. For example,
nucleophilic attack on silicon does not proceed by the
SN2 or
SN1
processes, but instead goes through a negatively charged true
pentacoordinate intermediate and appears like a substitution at a
hindered tertiary atom. This works for silicon, unlike for carbon,
because the long Si–C bonds reduce the steric hindrance and the
d-orbital of silicon is geometrically unconstrained for nucleophilic
attack, unlike for example a C–O σ* antibonding orbital. Nevertheless,
despite these differences, the mechanism is still often called "S
N2 at silicon" for simplicity.
[55]
One of the most useful silicon-containing groups is
trimethylsilyl, Me
3Si–.
The Si–C bond connecting it to the rest of the molecule is reasonably
strong, allowing it to remain while the rest of the molecule undergoes
reactions, but is not so strong that it cannot be removed specifically
when needed, for example by the
fluoride
ion, which is a very weak nucleophile for carbon compounds but a very
strong one for organosilicon compounds. It may be compared to acidic
protons;
while trisilylmethyl is removed by hard nucleophiles instead of bases,
both removals usually promote elimination. As a general rule, while
saturated carbon is best attacked by nucleophiles that are neutral
compounds, those based on nonmetals far down on the periodic table (e.g.
sulfur,
selenium, or
iodine),
or even both, silicon is best attacked by charged nucleophiles,
particularly those involving such highly electronegative nonmetals as
oxygen, fluorine, or chlorine. For example, enolates react at the carbon
in
haloalkanes, but at the oxygen in
silyl
chlorides; and when trimethylsilyl is removed from an organic molecule
using hydroxide as a nucleophile, the product of the reaction is not the
silanol as one would expect from using carbon chemistry as an analogy,
because the siloxide is strongly nucleophilic and attacks the original
molecule to yield the
silyl ether hexamethyldisiloxane, (Me
3Si)
2O. Conversely, while the S
N2 reaction is mostly unaffected by the presence of a partial positive charge (δ+) at the carbon, the analogous "S
N2" reaction at silicon is so affected. Thus, for example, the silyl
triflates are so electrophilic that they react 10
8 to 10
9 times faster than silyl chlorides with oxygen-containing nucleophiles.
Trimethylsilyl triflate is in particular a very good
Lewis acid and is used to convert
carbonyl compounds to
acetals and
silyl enol ethers, reacting them together analogously to the
aldol reaction.
[55]
Si–C bonds are commonly formed in three ways. In the laboratory,
preparation is often carried out in small quantities by reacting
tetrachlorosilane with
organolithium,
Grignard, or
organoaluminium
reagents, or by catalytic addition of Si–H across C=C double bonds. The
second route has the drawback of not being applicable to the most
important silanes, the methyl and phenyl silanes. Organosilanes are made
industrially by directly reacting alkyl or aryl halides with silicon
with 10% by weight metallic
copper
as a catalyst. Standard organic reactions suffice to produce many
derivatives; the resulting organosilanes are often significantly more
reactive than their carbon congeners, readily undergoing hydrolysis,
ammonolysis, alcoholysis, and condensation to form cyclic oligomers or
linear polymers.
[54]
Silicone polymers
The word "silicone" was first used by
Frederick Kipping in 1901. He invented the word to illustrate the similarity of chemical formulae between Ph
2SiO and
benzophenone, Ph
2CO, although he also stressed the lack of chemical resemblance due to the polymeric structure of Ph
2SiO, which is not shared by Ph
2CO.
[54]
Silicones may be considered analogous to mineral silicates, in which the methyl groups of the silicones correspond to the
isoelectronic O
− of the silicates.
[54]
They are quite stable to extreme temperatures, oxidation, and water,
and have useful dielectric, antistick, and antifoam properties.
Furthermore, they are resistant over long periods of time to ultraviolet
radiation and weathering, and are inert physiologically. They are
fairly unreactive, but do react with concentrated solutions bearing the
hydroxide ion and fluorinating agents, and occasionally can be even used
as mild reagents for selective syntheses. For example, (Me
3Si)
2O is valuable for the preparation of derivatives of
molybdenum and
tungsten oxyhalides, converting a
tungsten hexachloride suspension in
dichloroethane solution quantitatively to WOCl
4 in under an hour at room temperature, and then to yellow WO
2Cl
2 at 100 °C in light petroleum at a yield of 95% overnight.
[54]
Occurrence
In the universe, silicon is the seventh most abundant element, coming after
hydrogen,
helium,
carbon,
nitrogen,
oxygen, and
neon.
These abundances are not replicated well on Earth due to substantial
separation of the elements taking place during the formation of the
Solar System.
Silicon makes up 27.2% of the Earth's crust by weight, second only to
oxygen at 45.5%, with which it is always associated in nature. Further
fractionation took place in the formation of the Earth by
planetary differentiation:
Earth's core, which makes up 31.5% of the mass of the Earth, has approximate composition Fe
25Ni
2Co
0.1S
3; the
mantle makes up 68.1% of the Earth's mass and is composed mostly of denser oxides and silicates, an example being
olivine, (Mg,Fe)
2SiO
4; while the lighter siliceous minerals such as
aluminosilicates rise to the surface and form the crust, making up 0.4% of the Earth's mass.
[56]
The crystallisation of
igneous rocks
from magma depends on a number of factors; among them are the chemical
composition of the magma, the cooling rate, and some properties of the
individual minerals to be formed, such as
lattice energy, melting point, and complexity of their crystal structure. As magma is cooled,
olivine appears first, followed by
pyroxene,
amphibole,
biotite mica,
orthoclase feldspar,
muscovite mica,
quartz,
zeolites,
and finally hydrothermal minerals. This sequence shows a trend towards
increasingly complex silicate units with cooling, and the introduction
of
hydroxide and
fluoride anions in addition to oxides. Many metals can substitute for silicon. After these igneous rocks undergo
weathering, transport, and deposition,
sedimentary rocks like clay, shale, and sandstone are formed.
Metamorphism also can occur at high temperatures and pressures, creating an even vaster variety of minerals.
[56]
Production
Silicon of 96–99% purity is made by reducing
quartzite or sand with highly pure
coke. The reduction is carried out in an
electric arc furnace, with an excess of SiO
2 used to stop
silicon carbide (SiC) from accumulating:
[30]
- SiO2 + 2 C → Si + 2 CO
- 2 SiC + SiO2 → 3 Si + 2 CO
This reaction, known as carbothermal reduction of silicon dioxide, is
usually conducted in the presence of scrap iron with low amounts of
phosphorus and
sulfur, produing
ferrosilicon.
[30] Ferrosilicon, an iron-silicon alloy that contains varying ratios of
elemental silicon and iron, accounts for about 80% of the world's
production of elemental silicon, with China, the leading supplier of
elemental silicon, providing 4.6 million
tonnes
(or 2/3 of the world output) of silicon, most of which is in the form
of ferrosilicon. It is followed by Russia (610,000 t), Norway (330,000
t), Brazil (240,000 t) and the United States (170,000 t).
[57]
Ferrosilicon is primarily used by the iron and steel industry (see
below) with primary use as alloying addition in iron or steel and for
de-oxidation of steel in integrated steel plants.
[30] Another sometimes used reaction is aluminothermal reduction of silicon dioxide, as follows:
[58]
- 3 SiO2 + 4 Al → 3 Si + 2 Al2O3
Leaching powdered 96–97% pure silicon with water results in ~98.5%
pure silicon, which is used in the chemical industry. However, even
greater purity is needed for semiconductor applications, and this is
produced from the reduction of tetrachlorosilane or trichlorosilane. The
former is made by chlorinating scrap silicon and the latter is a
byproduct of
silicone production. These compounds are volatile and hence can be purified by repeated
fractional distillation, followed by reduction to elemental silicon with very pure
zinc
metal as the reducing agent. The spongy pieces of silicon thus produced
are melted and then grown to form cylindrical single crystals, before
being purified by
zone refining. Other routes use the thermal decomposition of silane or tetraiodosilane. Another process used is the reduction of
sodium hexafluorosilicate, a common waste product of the phosphate fertiliser industry, by metallic
sodium:
this is highly exothermic and hence requires no outside fuel source.
Hyperfine silicon is made at a higher purity than almost every other
material:
transistor production requires impurity levels in silicon crystals less than 1 part per 10
10, and in special cases impurity levels below 1 part per 10
12 are needed and attained.
[30]
Applications
Compounds
Most silicon is used industrially without being purified, and indeed
often with comparatively little processing from its natural form. Over
90% of the Earth's crust is composed of
silicate minerals,
which are compounds of silicon and oxygen, often with metallic ions
when negatively charged silicate anions require cations to balance the
charge. Many of these have direct commercial uses, such as clays,
silica
sand and most kinds of building stone. Thus, the vast majority of uses
for silicon are as structural compounds, either as the silicate minerals
or silica (crude silicon dioxide). Silicates are used in making
Portland cement (made mostly of calcium silicates) which is used in
building mortar and modern
stucco, but more importantly, combined with silica sand, and gravel (usually containing silicate minerals like granite), to make the
concrete that is the basis of most of the very largest industrial building projects of the modern world.
[59]
Silica is used to make
fire brick, a type of ceramic. Silicate minerals are also in whiteware
ceramics, an important class of products usually containing various types of fired
clay minerals (natural aluminium phyllosilicates). An example is
porcelain which is based on the silicate mineral
kaolinite. Traditional
glass (silica-based
soda-lime glass) also functions in many of the same ways, and is also used for windows and containers. In addition, specialty silica based
glass fibers are used for
optical fiber, as well as to produce
fiberglass for structural support and
glass wool for thermal insulation.
Silicones are often used in
waterproofing treatments,
molding compounds, mold-
release agents, mechanical seals, high temperature
greases and waxes, and
caulking compounds. Silicone is also sometimes used in
breast implants, contact lenses,
explosives and
pyrotechnics.
[60] Silly Putty was originally made by adding
boric acid to
silicone oil.
[61] Other silicon compounds function as high-technology abrasives and new high-strength ceramics based upon
silicon carbide. Silicon is a component of some
superalloys.
Alloys
Elemental silicon is added to molten
cast iron as
ferrosilicon or silicocalcium alloys to improve performance in casting thin sections and to prevent the formation of
cementite
where exposed to outside air. The presence of elemental silicon in
molten iron acts as a sink for oxygen, so that the steel carbon content,
which must be kept within narrow limits for each type of steel, can be
more closely controlled. Ferrosilicon production and use is a monitor of
the steel industry, and although this form of elemental silicon is
grossly impure, it accounts for 80% of the world's use of free silicon.
Silicon is an important constituent of
electrical steel, modifying its
resistivity and
ferromagnetic properties.
The properties of silicon can be used to modify alloys with metals
other than iron. "Metallurgical grade" silicon is silicon of 95–99%
purity. About 55% of the world consumption of metallurgical purity
silicon goes for production of aluminium-silicon alloys (
silumin alloys) for aluminium part
casts, mainly for use in the
automotive industry. Silicon's importance in aluminium casting is that a significantly high amount (12%) of silicon in aluminium forms a
eutectic mixture
which solidifies with very little thermal contraction. This greatly
reduces tearing and cracks formed from stress as casting alloys cool to
solidity. Silicon also significantly improves the hardness and thus
wear-resistance of aluminium.
[62][63]
Electronics

Silicon wafer with mirror finish
Most elemental silicon produced remains as a ferrosilicon alloy, and
only about 20% is refined to metallurgical grade purity (a total of
1.3–1.5 million metric tons/year). An estimated 15% of the world
production of metallurgical grade silicon is further refined to
semiconductor purity.
[63] This typically is the "nine-9" or 99.9999999% purity
[64] nearly defect-free single
crystalline material.
[65]
Monocrystalline silicon of such purity is usually produced by the
Czochralski process, is used to produce
silicon wafers used in the
semiconductor industry, in electronics, and in some high-cost and high-efficiency
photovoltaic applications.
[66] Pure silicon is an
intrinsic semiconductor, which means that unlike metals, it conducts
electron holes and electrons released from atoms by heat; silicon's
electrical conductivity increases with higher temperatures. Pure silicon has too low a conductivity (i.e., too high a
resistivity) to be used as a circuit element in electronics. In practice, pure silicon is
doped
with small concentrations of certain other elements, which greatly
increase its conductivity and adjust its electrical response by
controlling the number and charge (
positive or
negative) of activated carriers. Such control is necessary for
transistors,
solar cells,
semiconductor detectors, and other
semiconductor devices used in the computer industry and other technical applications.
[67] In
silicon photonics, silicon can be used as a continuous wave
Raman laser medium to produce coherent light.
[68]
In common
integrated circuits,
a wafer of monocrystalline silicon serves as a mechanical support for
the circuits, which are created by doping and insulated from each other
by thin layers of
silicon oxide,
an insulator that is easily produced by exposing the element to oxygen
under the proper conditions. Silicon has become the most popular
material for both high power semiconductors and integrated circuits
because it can withstand the highest temperatures and greatest
electrical activity without suffering
avalanche breakdown (an
electron avalanche
is created when heat produces free electrons and holes, which in turn
pass more current, which produces more heat). In addition, the
insulating oxide of silicon is not soluble in water, which gives it an
advantage over
germanium (an element with similar properties which can also be used in semiconductor devices) in certain fabrication techniques.
[69]
Monocrystalline silicon is expensive to produce, and is usually
justified only in production of integrated circuits, where tiny crystal
imperfections can interfere with tiny circuit paths. For other uses,
other types of pure silicon may be employed. These include
hydrogenated amorphous silicon and upgraded metallurgical-grade silicon (UMG-Si) used in the production of low-cost,
large-area electronics in applications such as
liquid crystal displays and of large-area, low-cost, thin-film
solar cells.
Such semiconductor grades of silicon are either slightly less pure or
polycrystalline rather than monocrystalline, and are produced in
comparable quatities as the monocrystalline silicon: 75,000 to 150,000
metric tons per year. The market for the lesser grade is growing more
quickly than for monocrystalline silicon. By 2013, polycrystalline
silicon production, used mostly in solar cells, was projected to reach
200,000 metric tons per year, while monocrystalline semiconductor grade
silicon was expected to remain less than 50,000 tons/year.
[63]
Biological role
A diatom, enclosed in a silica cell wall
Although silicon is readily available in the form of
silicates, very few organisms use it directly.
Diatoms,
radiolaria and
siliceous sponges use
biogenic silica as a structural material for skeletons. In more advanced plants, the silica
phytoliths (opal phytoliths) are rigid microscopic bodies occurring in the cell; some plants, for example
rice, need silicon for their growth.
[70][71][72] There is some evidence that silicon is important to nail, hair, bone and skin health in humans,
[73] for example in studies that show that premenopausal women with higher dietary silicon intake have higher
bone density, and that silicon supplementation can increase bone volume and density in patients with
osteoporosis.
[74] Silicon is needed for synthesis of
elastin and
collagen, of which the
aorta contains the greatest quantity in the human body
[75] and has been considered an
essential element;
[76]
nevertheless, it is difficult to prove its essentiality, because
silicon is very common and hence deficiency symptoms are difficult to
reproduce.
[77]
Silicon is currently under consideration for elevation to the status
of a "plant beneficial substance by the Association of American Plant
Food Control Officials (AAPFCO)."
[78][79] Silicon has been shown to improve plant cell wall strength and structural integrity in some plants.
[80]
Safety
People can be exposed to elemental silicon in the workplace by
breathing it in, swallowing it, skin contact, and eye contact. In the
latter two cases, silicon poses a slight hazard as an irritant; it is
hazardous if inhaled.
[81] The
Occupational Safety and Health Administration (OSHA) has set the legal limit (
Permissible exposure limit) for silicon exposure in the workplace as 15 mg/m
3 total exposure and 5 mg/m
3 respiratory exposure over an 8-hour workday. The
National Institute for Occupational Safety and Health (NIOSH) has set a
Recommended exposure limit (REL) of 10 mg/m
3 total exposure and 5 mg/m
3 respiratory exposure over an 8-hour workday.
[82] Inhalation of
crystalline silica dust may lead to
silicosis, an
occupational lung disease marked by
inflammation and scarring in the form of
nodular lesions in the upper lobes of the
lungs.
[83]




















.JPG)















