| Cytochrome P450 | |
|---|---|
.png)
Structure of lanosterol 14α-demethylase (CYP51)
|
Cytochromes P450 (CYPs) are proteins of the superfamily containing heme as a cofactor and, therefore, are hemeproteins. CYPs use a variety of small and large molecules as substrates in enzymatic reactions. They are, in general, the terminal oxidase enzymes in electron transfer chains, broadly categorized as P450-containing systems. The term "P450" is derived from the spectrophotometric peak at the wavelength of the absorption maximum of the enzyme (450 nm) when it is in the reduced state and complexed with carbon monoxide.
CYP enzymes have been identified in all kingdoms of life: animals, plants, fungi, protists, bacteria, archaea, and even in viruses. However, they are not omnipresent; for example, they have not been found in Escherichia coli. More than 50,000 distinct CYP proteins are known.
Most CYPs require a protein partner to deliver one or more electrons to reduce the iron (and eventually molecular oxygen). Based on the nature of the electron transfer proteins, CYPs can be classified into several groups:
- Microsomal P450 systems, in which electrons are transferred from NADPH via cytochrome P450 reductase (variously CPR, POR, or CYPOR). Cytochrome b5 (cyb5) can also contribute reducing power to this system after being reduced by cytochrome b5 reductase (CYB5R).
- Mitochondrial P450 systems, which employ adrenodoxin reductase and adrenodoxin to transfer electrons from NADPH to P450.
- Bacterial P450 systems, which employ a ferredoxin reductase and a ferredoxin to transfer electrons to P450.
- CYB5R/cyb5/P450 systems, in which both electrons required by the CYP come from cytochrome b5.
- FMN/Fd/P450 systems, originally found in Rhodococcus species, in which a FMN-domain-containing reductase is fused to the CYP.
- P450 only systems, which do not require external reducing power. Notable ones include thromboxane synthase (CYP5), prostacyclin synthase (CYP8), and CYP74A (allene oxide synthase).
Many hydroxylation reactions (insertion of hydroxyl groups) use CYP enzymes.
Nomenclature
Genes encoding CYP enzymes, and the enzymes themselves, are designated with the root symbol CYP for the superfamily, followed by a number indicating the gene family, a capital letter indicating the subfamily, and another numeral for the individual gene. The convention is to italicise the name when referring to the gene. For example, CYP2E1 is the gene that encodes the enzyme CYP2E1—one of the enzymes involved in paracetamol (acetaminophen) metabolism. The CYP nomenclature is the official naming convention, although occasionally CYP450 or CYP450 is used synonymously. However, some gene or enzyme names for CYPs may differ from this nomenclature, denoting the catalytic activity and the name of the compound used as substrate. Examples include CYP5A1, thromboxane A2 synthase, abbreviated to TBXAS1 (ThromBoXane A2 Synthase 1), and CYP51A1, lanosterol 14-α-demethylase, sometimes unofficially abbreviated to LDM according to its substrate (Lanosterol) and activity (DeMethylation).The current nomenclature guidelines suggest that members of new CYP families share at least 40% amino acid identity, while members of subfamilies must share at least 55% amino acid identity. There are nomenclature committees that assign and track both base gene names (Cytochrome P450 Homepage) and allele names (CYP Allele Nomenclature Committee).
Mechanism

The "Fe(V) intermediate" at the bottom left is a simplification: it is an Fe(IV) with a radical heme ligand.
Structure
The active site of cytochrome P450 contains a heme-iron center. The iron is tethered to the protein via a cysteine thiolate ligand. This cysteine and several flanking residues are highly conserved in known CYPs and have the formal PROSITE signature consensus pattern [FW] - [SGNH] - x - [GD] - {F} - [RKHPT] - {P} - C - [LIVMFAP] - [GAD]. Because of the vast variety of reactions catalyzed by CYPs, the activities and properties of the many CYPs differ in many aspects. In general, the P450 catalytic cycle proceeds as follows:Catalytic cycle
- Substrate binds in proximity to the heme group, on the side opposite to the axial thiolate. Substrate binding induces a change in the conformation of the active site, often displacing a water molecule from the distal axial coordination position of the heme iron, and changing the state of the heme iron from low-spin to high-spin.
- Substrate binding induces electron transfer from NAD(P)H via cytochrome P450 reductase or another associated reductase.
- Molecular oxygen binds to the resulting ferrous heme center at the distal axial coordination position, initially giving a dioxygen adduct not unlike oxy-myoglobin.
- A second electron is transferred, from either cytochrome P450 reductase, ferredoxins, or cytochrome b5, reducing the Fe-O2 adduct to give a short-lived peroxo state.
- The peroxo group formed in step 4 is rapidly protonated twice, releasing one molecule of water and forming the highly reactive species referred to as P450 Compound 1 (or just Compound I). This highly reactive intermediate was isolated in 2010, P450 Compound 1 is an iron(IV) oxo (or ferryl) species with an additional oxidizing equivalent delocalized over the porphyrin and thiolate ligands. Evidence for the alternative perferryl iron(V)-oxo is lacking.
- Depending on the substrate and enzyme involved, P450 enzymes can catalyze any of a wide variety of reactions. A hypothetical hydroxylation is shown in this illustration. After the product has been released from the active site, the enzyme returns to its original state, with a water molecule returning to occupy the distal coordination position of the iron nucleus.
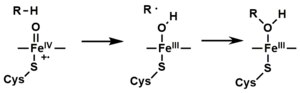
Oxygen rebound mechanism
utilized by cytochrome P450 for conversion of hydrocarbons to alcohols
via the action of "compound I", an iron(IV) oxide bound to a heme
radical cation.
- An alternative route for mono-oxygenation is via the "peroxide shunt" (path "S" in figure). This pathway entails oxidation of the ferric-substrate complex with oxygen-atom donors such as peroxides and hypochlorites. A hypothetical peroxide "XOOH" is shown in the diagram.
Spectroscopy
Binding of substrate is reflected in the spectral properties of the enzyme, with an increase in absorbance at 390 nm and a decrease at 420 nm. This can be measured by difference spectrometry and is referred to as the "type I" difference spectrum (see inset graph in figure). Some substrates cause an opposite change in spectral properties, a "reverse type I" spectrum, by processes that are as yet unclear. Inhibitors and certain substrates that bind directly to the heme iron give rise to the type II difference spectrum, with a maximum at 430 nm and a minimum at 390 nm (see inset graph in figure). If no reducing equivalents are available, this complex may remain stable, allowing the degree of binding to be determined from absorbance measurements in vitro C: If carbon monoxide (CO) binds to reduced P450, the catalytic cycle is interrupted. This reaction yields the classic CO difference spectrum with a maximum at 450 nm.P450s in humans
Human CYPs are primarily membrane-associated proteins located either in the inner membrane of mitochondria or in the endoplasmic reticulum of cells. CYPs metabolize thousands of endogenous and exogenous chemicals. Some CYPs metabolize only one (or a very few) substrates, such as CYP19 (aromatase), while others may metabolize multiple substrates. Both of these characteristics account for their central importance in medicine. Cytochrome P450 enzymes are present in most tissues of the body, and play important roles in hormone synthesis and breakdown (including estrogen and testosterone synthesis and metabolism), cholesterol synthesis, and vitamin D metabolism. Cytochrome P450 enzymes also function to metabolize potentially toxic compounds, including drugs and products of endogenous metabolism such as bilirubin, principally in the liver.The Human Genome Project has identified 57 human genes coding for the various cytochrome P450 enzymes.
Drug metabolism

Proportion of antifungal drugs metabolized by different families of CYPs.
CYPs are the major enzymes involved in drug metabolism, accounting for about 75% of the total metabolism. Most drugs undergo deactivation by CYPs, either directly or by facilitated excretion from the body. Also, many substances are bioactivated by CYPs to form their active compounds.
Drug interaction
Many drugs may increase or decrease the activity of various CYP isozymes either by inducing the biosynthesis of an isozyme (enzyme induction) or by directly inhibiting the activity of the CYP (enzyme inhibition). This is a major source of adverse drug interactions, since changes in CYP enzyme activity may affect the metabolism and clearance of various drugs. For example, if one drug inhibits the CYP-mediated metabolism of another drug, the second drug may accumulate within the body to toxic levels. Hence, these drug interactions may necessitate dosage adjustments or choosing drugs that do not interact with the CYP system. Such drug interactions are especially important to take into account when using drugs of vital importance to the patient, drugs with important side-effects and drugs with small therapeutic windows, but any drug may be subject to an altered plasma concentration due to altered drug metabolism.A classical example includes anti-epileptic drugs. Phenytoin, for example, induces CYP1A2, CYP2C9, CYP2C19, and CYP3A4. Substrates for the latter may be drugs with critical dosage, like amiodarone or carbamazepine, whose blood plasma concentration may either increase because of enzyme inhibition in the former, or decrease because of enzyme induction in the latter.
Interaction of other substances
Naturally occurring compounds may also induce or inhibit CYP activity. For example, bioactive compounds found in grapefruit juice and some other fruit juices, including bergamottin, dihydroxybergamottin, and paradicin-A, have been found to inhibit CYP3A4-mediated metabolism of certain medications, leading to increased bioavailability and, thus, the strong possibility of overdosing. Because of this risk, avoiding grapefruit juice and fresh grapefruits entirely while on drugs is usually advised.Other examples:
- Saint-John's wort, a common herbal remedy induces CYP3A4, but also inhibits CYP1A1, CYP1B1.
- Tobacco smoking induces CYP1A2 (example CYP1A2 substrates are clozapine, olanzapine, and fluvoxamine)
- At relatively high concentrations, starfruit juice has also been shown to inhibit CYP2A6 and other CYPs. Watercress is also a known inhibitor of the cytochrome P450 CYP2E1, which may result in altered drug metabolism for individuals on certain medications (e.g., chlorzoxazone).
- Tributyltin has been found to inhibit the function of cytochrome P450, leading to masculinization of mollusks.
- Goldenseal, with its two notable alkaloids berberine and hydrastine, has been shown to alter P450-marker enzymatic activities (involving CYP2C9, CYP2D6, and CYP3A4).
Other specific CYP functions
Steroid hormones

Steroidogenesis, showing many of the enzyme activities that are performed by cytochrome P450 enzymes. HSD: Hydroxysteroid dehydrogenase.
A subset of cytochrome P450 enzymes play important roles in the synthesis of steroid hormones (steroidogenesis) by the adrenals, gonads, and peripheral tissue:
- CYP11A1 (also known as P450scc or P450c11a1) in adrenal mitochondria affects "the activity formerly known as 20,22-desmolase" (steroid 20α-hydroxylase, steroid 22-hydroxylase, cholesterol side-chain scission).
- CYP11B1 (encoding the protein P450c11β) found in the inner mitochondrial membrane of adrenal cortex has steroid 11β-hydroxylase, steroid 18-hydroxylase, and steroid 18-methyloxidase activities.
- CYP11B2 (encoding the protein P450c11AS), found only in the mitochondria of the adrenal zona glomerulosa, has steroid 11β-hydroxylase, steroid 18-hydroxylase, and steroid 18-methyloxidase activities.
- CYP17A1, in endoplasmic reticulum of adrenal cortex has steroid 17α-hydroxylase and 17,20-lyase activities.
- CYP21A1 (P450c21) in adrenal cortex conducts 21-hydroxylase activity.
- CYP19A (P450arom, aromatase) in endoplasmic reticulum of gonads, brain, adipose tissue, and elsewhere catalyzes aromatization of androgens to estrogens.
Polyunsaturated fatty acids and eicosanoids
Certain cytochrome P450 enzymes are critical in metabolizing polyunsaturated fatty acids (PUFAs) to biologically active, intercellular cell signaling molecules (eicosanoids) and/or metabolize biologically active metabolites of the PUFA to less active or inactive products. These CYPs possess cytochrome P450 omega hydroxylase and/or epoxygenase enzyme activity.- CYP1A1, CYP1A2, and CYP2E1 metabolize endogenous PUFAs to signaling molecules: they metabolize arachidonic acid (i.e. AA) to 19-hydroxyeicosatetraenoic acid (i.e. 19-HETE; see 20-hydroxyeicosatetraenoic acid); eicosapentaenoic acid (i.e. EPA) to epoxyeicosatetraenoic acids (i.e. EEQs); and docosahexaenoic acid (i.e. DHA) to epoxydocosapentaenoic acids (i.e. EDPs).
- CYP2C8, CYP2C9, CYP2C18, CYP2C19, and CYP2J2 metabolize endogenous PUFAs to signaling molecules: they metabolize AA to epoxyeicosatetraenoic acids (i.e. EETs); EPA to EEQs; and DHA to EDPs.
- CYP2S1 metabolizes PUFA to signaling molecules: it metabolizes AA to EETs ad EPA to EEQs.
- CYP3A4 metabolizes AA to EET signaling molecules.
- CYP4A11 metabolizes endogenous PUFAs to signaling molecules: it metabolizes AA to 20-HETE and EETs; it also hydroxylates DHA to 22-hydroxy-DHA (i.e. 12-HDHA).
- CYP4F2, CYP4F3A, and CYP4F3B (see CYP4F3 for latter two CYPs) metabolize PUFAs to signaling molecules: they metabolizes AA to 20-HETE. They also metabolize EPA to 19-hydroxyeicosapentaenoic acid (19-HEPE) and 20-hydroxyeicosapentaenoic acid (20-HEPE) as well as metabolize DHA to 22-HDA. They also inactivate or reduce the activity of signaling molecules: they metabolize leukotriene B4 (LTB4) to 20-hydroxy-LTB4, 5-hydroxyeicosatetraenoic acid (5-HETE) to 5,20-diHETE, 5-oxo-eicosatetraenoic acid (5-oxo-ETE) to 5-oxo,20-hydroxy-ETE, 12-hydroxyeicosatetraenoic acid (12-HETE) to 12,20-diHETE, EETs to 20-hydroxy-EETs, and lipoxins to 20-hydroxy products.
- CYP4F8 and CYP4F12 metabolize PUFAs to signaling molecules: they metabolizes EPA to EEQs and DHA to EDPs. They also metabolize AA to 18-hydroxyeicosatetraenoic acid (18-HETE) and 19-HETE.
- CYP4F11 inactivates or reduces the activity of signaling molecules: it metabolizes LTB4 to 20-hydroxy-LTB4, (5-HETE) to 5,20-diHETE, (5-oxo-ETE) to 5-oxo,20-hydroxy-ETE, (12-HETE) to 12,20-diHETE, EETs to 20-hydroxy-EETs, and lipoxins to 20-hydroxy products.
- CYP4F22 ω-hydroxylates extremely long "very long chain fatty acids", i.e. fatty acids that are 28 or more carbons long. The ω-hydroxylation of these special fatty acids is critical to creating and maintaining the skins water barrier function; autosomal recessive inactivating mutations of CYP4F22 are associated with the Lamellar ichthyosis subtype of Congenital ichthyosiform erythrodema in humans.
CYP families in humans
Humans have 57 genes and more than 59 pseudogenes divided among 18 families of cytochrome P450 genes and 43 subfamilies. This is a summary of the genes and of the proteins they encode.| Family | Function | Members | Names |
| CYP1 | drug and steroid (especially estrogen) metabolism, benzo[a]pyrene toxification (forming (+)-benzo[a]pyrene-7,8-dihydrodiol-9,10-epoxide) | 3 subfamilies, 3 genes, 1 pseudogene | CYP1A1, CYP1A2, CYP1B1 |
| CYP2 | drug and steroid metabolism | 13 subfamilies, 16 genes, 16 pseudogenes | CYP2A6, CYP2A7, CYP2A13, CYP2B6, CYP2C8, CYP2C9, CYP2C18, CYP2C19, CYP2D6, CYP2E1, CYP2F1, CYP2J2, CYP2R1, CYP2S1, CYP2U1, CYP2W1 |
| CYP3 | drug and steroid (including testosterone) metabolism | 1 subfamily, 4 genes, 2 pseudogenes | CYP3A4, CYP3A5, CYP3A7, CYP3A43 |
| CYP4 | arachidonic acid or fatty acid metabolism | 6 subfamilies, 12 genes, 10 pseudogenes | CYP4A11, CYP4A22, CYP4B1, CYP4F2, CYP4F3, CYP4F8, CYP4F11, CYP4F12, CYP4F22, CYP4V2, CYP4X1, CYP4Z1 |
| CYP5 | thromboxane A2 synthase | 1 subfamily, 1 gene | CYP5A1 |
| CYP7 | bile acid biosynthesis 7-alpha hydroxylase of steroid nucleus | 2 subfamilies, 2 genes | CYP7A1, CYP7B1 |
| CYP8 | varied | 2 subfamilies, 2 genes | CYP8A1 (prostacyclin synthase), CYP8B1 (bile acid biosynthesis) |
| CYP11 | steroid biosynthesis | 2 subfamilies, 3 genes | CYP11A1, CYP11B1, CYP11B2 |
| CYP17 | steroid biosynthesis, 17-alpha hydroxylase | 1 subfamily, 1 gene | CYP17A1 |
| CYP19 | steroid biosynthesis: aromatase synthesizes estrogen | 1 subfamily, 1 gene | CYP19A1 |
| CYP20 | unknown function | 1 subfamily, 1 gene | CYP20A1 |
| CYP21 | steroid biosynthesis | 2 subfamilies, 1 gene, 1 pseudogene | CYP21A2 |
| CYP24 | vitamin D degradation | 1 subfamily, 1 gene | CYP24A1 |
| CYP26 | retinoic acid hydroxylase | 3 subfamilies, 3 genes | CYP26A1, CYP26B1, CYP26C1 |
| CYP27 | varied | 3 subfamilies, 3 genes | CYP27A1 (bile acid biosynthesis), CYP27B1 (vitamin D3 1-alpha hydroxylase, activates vitamin D3), CYP27C1 (unknown function) |
| CYP39 | 7-alpha hydroxylation of 24-hydroxycholesterol | 1 subfamily, 1 gene | CYP39A1 |
| CYP46 | cholesterol 24-hydroxylase | 1 subfamily, 1 gene | CYP46A1 |
| CYP51 | cholesterol biosynthesis | 1 subfamily, 1 gene, 3 pseudogenes | CYP51A1 (lanosterol 14-alpha demethylase) |
P450s in other species
Animals
Many animals have as many or more CYP genes than humans do. Reported numbers range from 35 genes in the sponge Amphimedon queenslandica to 235 genes in the cephalochordate Branchiostoma floridae. Mice have genes for 101 CYPs, and sea urchins have even more (perhaps as many as 120 genes). Most CYP enzymes are presumed to have monooxygenase activity, as is the case for most mammalian CYPs that have been investigated (except for, e.g., CYP19 and CYP5). Gene and genome sequencing is far outpacing biochemical characterization of enzymatic function, though many genes with close homology to CYPs with known function have been found, giving clues to their functionality.The classes of CYPs most often investigated in non-human animals are those either involved in development (e.g., retinoic acid or hormone metabolism) or involved in the metabolism of toxic compounds (such as heterocyclic amines or polyaromatic hydrocarbons). Often there are differences in gene regulation or enzyme function of CYPs in related animals that explain observed differences in susceptibility to toxic compounds (ex. canines inability to metabolize xanthines such as caffeine). Some drugs undergo metabolism in both species via different enzymes, resulting in different metabolites, while other drugs are metabolized in one species but excreted unchanged in another species. For this reason, one species's reaction to a substance is not a reliable indication of the substance's effects in humans.
CYPs have been extensively examined in mice, rats, dogs, and less so in zebrafish, in order to facilitate use of these model organisms in drug discovery and toxicology. Recently CYPs have also been discovered in avian species, in particular turkeys, that may turn out to be a useful model for cancer research in humans. CYP1A5 and CYP3A37 in turkeys were found to be very similar to the human CYP1A2 and CYP3A4 respectively, in terms of their kinetic properties as well as in the metabolism of aflatoxin B1.
CYPs have also been heavily studied in insects, often to understand pesticide resistance. For example, CYP6G1 is linked to insecticide resistance in DDT-resistant Drosophila melanogaster and CYP6Z1 in the mosquito malaria vector Anopheles gambiae is capable of directly metabolizing DDT.
Microbial
Microbial cytochromes P450 are often soluble enzymes and are involved in diverse metabolic processes. In bacteria the distribution of P450s is very variable with many bacteria having no identified P450s (e.g. E.coli). Some bacteria, predominantly actinomycetes, have numerous P450s (e.g.,). Those so far identified are generally involved in either biotransformation of xenobiotic compounds (e.g. CYP105A1 from Streptomyces griseolus metabolizes sulfonylurea herbicides to less toxic derivatives,) or are part of specialised metabolite biosynthetic pathways (e.g. CYP170B1 catalyses production of the sesquiterpenoid albaflavenone in Streptomyces albus,). Although no P450 has yet been shown to be essential in a microbe, the CYP105 family is highly conserved with a representative in every streptomycete genome sequenced so far. Due to the solubility of bacterial P450 enzymes, they are generally regarded as easier to work with than the predominantly membrane bound eukaryotic P450s. This, combined with the remarkable chemistry they catalyse, has led to many studies using the heterologously expressed proteins in vitro. Few studies have investigated what P450s do in vivo, what the natural substrate(s) are and how P450s contribute to survival of the bacteria in the natural environment.Three examples that have contributed significantly to structural and mechanistic studies are listed here, but many different families exist.- Cytochrome P450cam (CYP101) originally from Pseudomonas putida has been used as a model for many cytochromes P450 and was the first cytochrome P450 three-dimensional protein structure solved by X-ray crystallography. This enzyme is part of a camphor-hydroxylating catalytic cycle consisting of two electron transfer steps from putidaredoxin, a 2Fe-2S cluster-containing protein cofactor.
- Cytochrome P450 eryF (CYP107A1) originally from the actinomycete bacterium Saccharopolyspora erythraea is responsible for the biosynthesis of the antibiotic erythromycin by C6-hydroxylation of the macrolide 6-deoxyerythronolide B.
- Cytochrome P450 BM3 (CYP102A1) from the soil bacterium Bacillus megaterium catalyzes the NADPH-dependent hydroxylation of several long-chain fatty acids at the ω–1 through ω–3 positions. Unlike almost every other known CYP (except CYP505A1, cytochrome P450 foxy), it constitutes a natural fusion protein between the CYP domain and an electron donating cofactor. Thus, BM3 is potentially very useful in biotechnological applications.
- Cytochrome P450 119 (CYP119) isolated from the thermophillic archea Sulfolobus acidocaldarius has been used in a variety of mechanistic studies. Because thermophillic enzymes evolved to function at high temperatures, they tend to function more slowly at room temperature (if at all) and are therefore excellent mechanistic models.
Fungi
The commonly used azole class antifungal drugs work by inhibition of the fungal cytochrome P450 14α-demethylase. This interrupts the conversion of lanosterol to ergosterol, a component of the fungal cell membrane. (This is useful only because humans' P450 have a different sensitivity; this is how this class of antifungals work.)Significant research is ongoing into fungal P450s, as a number of fungi are pathogenic to humans (such as Candida yeast and Aspergillus) and to plants.
Cunninghamella elegans is a candidate for use as a model for mammalian drug metabolism.
Plants
Plant cytochrome P450s are involved in a wide range of biosynthetic reactions and target a diverse range of biomolecules. These reactions lead to various fatty acid conjugates, plant hormones, secondary metabolites, lignins, and a variety of defensive compounds. Plant genome annotations suggest that cytochrome P450 genes make up as much as 1% of the plant genes. The number and diversity of P450 genes is responsible, in part, for the multitude of bioactive compounds.P450s in biotechnology
The remarkable reactivity and substrate promiscuity of P450s have long attracted the attention of chemists. Recent progress towards realizing the potential of using P450s towards difficult oxidations have included: (i) eliminating the need for natural co-factors by replacing them with inexpensive peroxide containing molecules, (ii) exploring the compatibility of P450s with organic solvents, and (iii) the use of small, non-chiral auxiliaries to predictably direct P450 oxidation.InterPro subfamilies
InterPro subfamilies:- Cytochrome P450, B-class InterPro: IPR002397
- Cytochrome P450, mitochondrial InterPro: IPR002399
- Cytochrome P450, E-class, group I InterPro: IPR002401
- Cytochrome P450, E-class, group II InterPro: IPR002402
- Cytochrome P450, E-class, group IV InterPro: IPR002403
- Aromatase
These enzymes are of interest, because in assays, they can activate compounds to carcinogens. High levels of CYP1A2 have been linked to an increased risk of colon cancer. Since the 1A2 enzyme can be induced by cigarette smoking, this links smoking with colon cancer.











