Metastasis is a pathogenic agent's spread from an initial or primary site to a different or secondary site within the host's body; the term is typically used when referring to metastasis by a cancerous tumor. The newly pathological sites, then, are metastases (mets). It is generally distinguished from cancer invasion, which is the direct extension and penetration by cancer cells into neighboring tissues.
Cancer occurs after cells are genetically altered to proliferate rapidly and indefinitely. This uncontrolled proliferation by mitosis produces a primaryheterogeneic tumour. The cells which constitute the tumor eventually undergo metaplasia, followed by dysplasia then anaplasia, resulting in a malignant phenotype. This malignancy allows for invasion into the circulation, followed by invasion to a second site for tumorigenesis.
Some cancer cells known as circulating tumor cells acquire the ability to penetrate the walls of lymphatic or blood vessels, after which they are able to circulate through the bloodstream to other sites and tissues in the body. This process is known (respectively) as lymphatic or hematogenous
spread. After the tumor cells come to rest at another site, they
re-penetrate the vessel or walls and continue to multiply, eventually
forming another clinically detectable tumor. This new tumor is known as a metastatic (or secondary) tumor. Metastasis is one of the hallmarks of cancer, distinguishing it from benign tumors. Most cancers can metastasize, although in varying degrees. Basal cell carcinoma for example rarely metastasizes.
When tumor cells metastasize, the new tumor is called a secondary or metastatic tumor, and its cells are similar to those in the original or primary tumor. This means that if breast cancer
metastasizes to the lungs, the secondary tumor is made up of abnormal
breast cells, not of abnormal lung cells. The tumor in the lung is then
called metastatic breast cancer, not lung cancer. Metastasis is a key element in cancer staging systems such as the TNM staging system, where it represents the "M". In overall stage grouping,
metastasis places a cancer in Stage IV. The possibilities of curative
treatment are greatly reduced, or often entirely removed when a cancer
has metastasized.
Signs and symptoms
Cut surface of a liver showing multiple paler metastatic nodules originating from pancreatic cancer
Initially, nearby lymph nodes are struck early. The lungs, liver, brain, and bones are the most common metastasis locations from solid tumors.
Although advanced cancer may cause pain, it is often not the first symptom.
Some patients, however, do not show any symptoms.
When the organ gets a metastatic disease it begins to shrink until its lymph nodes burst, or undergo lysis.
Pathophysiology
Metastatic
tumors are very common in the late stages of cancer. The spread of
metastasis may occur via the blood or the lymphatics or through both
routes. The most common sites of metastases are the lungs, liver, brain, and the bones.
Currently, three main theories have been proposed to explain the
metastatic pathway of cancer: the epithelial-mesenchymal transition
(EMT) and mesenchymal-epithelial transition (MET) hypothesis (1), the
cancer stem cell hypothesis (2), and the macrophage–cancer cell fusion
hybrid hypothesis (3). Some new hypotheses were suggested as well, i.e.,
under the effect of particular biochemical and/or physical stressors,
cancer cells can undergo nuclear expulsion with subsequent macrophage
engulfment and fusion, with the formation of cancer fusion cells (CFCs).
Factors involved
Metastasis
involves a complex series of steps in which cancer cells leave the
original tumor site and migrate to other parts of the body via the
bloodstream, via the lymphatic system, or by direct extension. To do so,
malignant cells break away from the primary tumor and attach to and
degrade proteins that make up the surrounding extracellular matrix
(ECM), which separates the tumor from adjoining tissues. By degrading
these proteins, cancer cells are able to breach the ECM and escape. The
location of the metastases is not always random, with different types of
cancer tending to spread to particular organs and tissues at a rate
that is higher than expected by statistical chance alone.
Breast cancer, for example, tends to metastasize to the bones and
lungs. This specificity seems to be mediated by soluble signal molecules
such as chemokines and transforming growth factor beta. The body resists metastasis by a variety of mechanisms through the actions of a class of proteins known as metastasis suppressors, of which about a dozen are known.
Human cells exhibit different kinds of motion: collective motility, mesenchymal-type movement, and amoeboid movement.
Cancer cells often opportunistically switch between different kinds of
motion. Some cancer researchers hope to find treatments that can stop or
at least slow down the spread of cancer by somehow blocking some
necessary step in one or more kinds of motion.
All steps of the metastatic cascade involve a number of physical
processes. Cell migration requires the generation of forces, and when
cancer cells transmigrate through the vasculature, this requires
physical gaps in the blood vessels to form. Besides forces, the regulation of various types of cell-cell and cell-matrix adhesions is crucial during metastasis.
The metastatic steps are critically regulated by various cell
types, including the blood vessel cells (endothelial cells), immune
cells or stromal cells. The growth of a new network of blood vessels,
called tumor angiogenesis, is a crucial hallmark of cancer. It has therefore been suggested that angiogenesis inhibitors would prevent the growth of metastases. Endothelial progenitor cells have been shown to have a strong influence on metastasis and angiogenesis. Endothelial progenitor cells are important in tumor growth, angiogenesis and metastasis, and can be marked using the Inhibitor of DNA Binding 1
(ID1). This novel finding meant that investigators gained the ability
to track endothelial progenitor cells from the bone marrow to the blood
to the tumor-stroma and even incorporated in tumor vasculature.
Endothelial progenitor cells incorporated in tumor vasculature suggests
that this cell type in blood-vessel development is important in a tumor
setting and metastasis. Furthermore, ablation of the endothelial
progenitor cells in the bone marrow can lead to a significant decrease
in tumor growth and vasculature development. Therefore, endothelial
progenitor cells are important in tumor biology and present novel
therapeutic targets. The immune system is typically deregulated in cancer and affects many stages of tumor progression, including metastasis.
Epigenetic
regulation also plays an important role in the metastatic outgrowth of
disseminated tumor cells. Metastases display alterations in histone
modifications, such as H3K4-methylation and H3K9-methylation, when
compared to matching primary tumors.
These epigenetic modifications in metastases may allow the
proliferation and survival of disseminated tumor cells in distant
organs.
A recent study shows that PKC-iota promotes melanoma cell
invasion by activating Vimentin during EMT. PKC-iota inhibition or
knockdown resulted in an increase in E-cadherin and RhoA levels while
decreasing total Vimentin, phosphorylated Vimentin (S39) and Par6 in
metastatic melanoma cells. These results suggested that PKC-ι is
involved in signaling pathways which upregulate EMT in melanoma thereby
directly stimulates metastasis.
Recently, a series of high-profile experiments suggests that the
co-option of intercellular cross-talk mediated by exosome vesicles is a
critical factor involved in all steps of the invasion-metastasis
cascade.
Routes
Metastasis occurs by the following four routes:
Transcoelomic
The spread of a malignancy into body cavities can occur via penetrating the surface of the peritoneal, pleural, pericardial, or subarachnoid spaces. For example, ovarian tumors can spread transperitoneally to the surface of the liver.
Lymphatic spread
Lymphatic spread allows the transport of tumor cells to regional lymph nodes
near the primary tumor and ultimately, to other parts of the body. This
is called nodal involvement, positive nodes, or regional disease.
"Positive nodes" is a term that would be used by medical specialists to
describe regional lymph nodes that tested positive for malignancy. It is
common medical practice to test by biopsy at least one lymph node near a
tumor site when carrying out surgery to examine or remove a tumor. This
lymph node is then called a sentinel lymph node.
Lymphatic spread is the most common route of initial metastasis for carcinomas. In contrast, it is uncommon for a sarcoma
to metastasize via this route. Localized spread to regional lymph nodes
near the primary tumor is not normally counted as a metastasis,
although this is a sign of a worse outcome.
The lymphatic system does eventually drain from the thoracic duct and right lymphatic duct into the systemic venous system at the venous angle and into the brachiocephalic veins, and therefore these metastatic cells can also eventually spread through the haematogenous route.
Lymph node with almost complete replacement by metastatic melanoma. The brown pigment is focal deposition of melanin
Hematogenous spread
This is typical route of metastasis for sarcomas, but it is also the favored route for certain types of carcinoma, such as renal cell carcinoma originating in the kidney
and follicular carcinomas of the thyroid. Because of their thinner
walls, veins are more frequently invaded than are arteries, and
metastasis tends to follow the pattern of venous flow. That is, hematogenous spread often follows distinct patterns depending on the location of the primary tumor. For example, colorectal cancer spreads primarily through the portal vein to the liver.
Canalicular spread
Some tumors, especially carcinomas
may metastasize along anatomical canalicular spaces. These spaces
include for example the bile ducts, the urinary system, the airways and
the subarachnoid space.
The process is similar to that of transcoelomic spread. However, often
it remains unclear whether simultaneously diagnosed tumors of a
canalicular system are one metastatic process or in fact independent
tumors caused by the same agent (field cancerization).
Organ-specific targets
Main sites of metastases for some common cancer types. Primary cancers are denoted by "...cancer" and their main metastasis sites are denoted by "...metastases".
There is a propensity for certain tumors to seed in particular
organs. This was first discussed as the "seed and soil" theory by Stephen Paget in 1889. The propensity for a metastatic cell to spread to a particular organ is termed 'organotropism'. For example, prostate cancer usually metastasizes to the bones. In a similar manner, colon cancer has a tendency to metastasize to the liver. Stomach cancer often metastasises to the ovary in women, when it is called a Krukenberg tumor.
According to the "seed and soil" theory, it is difficult for
cancer cells to survive outside their region of origin, so in order to
metastasize they must find a location with similar characteristics. For example, breast tumor cells, which gather calcium ions from breast milk, metastasize to bone tissue, where they can gather calcium ions from bone. Malignant melanoma spreads to the brain, presumably because neural tissue and melanocytes arise from the same cell line in the embryo.
In 1928, James Ewing
challenged the "seed and soil" theory and proposed that metastasis
occurs purely by anatomic and mechanical routes. This hypothesis has
been recently utilized to suggest several hypotheses about the life
cycle of circulating tumor cells (CTCs) and to postulate that the
patterns of spread could be better understood through a 'filter and
flow' perspective.
However, contemporary evidences indicate that the primary tumour may
dictate organotropic metastases by inducing the formation of pre-metastatic niches at distant sites, where incoming metastatic cells may engraft and colonise.
Specifically, exosome vesicles secreted by tumours have been shown to
home to pre-metastatic sites, where they activate pro-metastatic
processes such as angiogenesis and modify the immune contexture, so as
to foster a favourable microenvironment for secondary tumour growth.
Metastasis and primary cancer
It
is theorized that metastasis always coincides with a primary cancer,
and, as such, is a tumor that started from a cancer cell or cells in
another part of the body. However, over 10% of patients presenting to oncology units
will have metastases without a primary tumor found. In these cases,
doctors refer to the primary tumor as "unknown" or "occult," and the
patient is said to have cancer of unknown primary origin (CUP) or unknown primary tumors (UPT). It is estimated that 3% of all cancers are of unknown primary origin. Studies have shown that, if simple questioning does not reveal the cancer's source (coughing up blood—"probably lung", urinating blood—"probably bladder"), complex imaging will not either. In some of these cases a primary tumor may appear later.
The use of immunohistochemistry
has permitted pathologists to give an identity to many of these
metastases. However, imaging of the indicated area only occasionally
reveals a primary. In rare cases (e.g., of melanoma), no primary tumor is found, even on autopsy.
It is therefore thought that some primary tumors can regress
completely, but leave their metastases behind. In other cases, the tumor
might just be too small and/or in an unusual location to be diagnosed.
Diagnosis
Pulmonary metastases shown on Chest X-Ray
The cells in a metastatic tumor resemble those in the primary tumor.
Once the cancerous tissue is examined under a microscope to determine
the cell type, a doctor can usually tell whether that type of cell is
normally found in the part of the body from which the tissue sample was
taken.
For instance, breast cancer
cells look the same whether they are found in the breast or have spread
to another part of the body. So, if a tissue sample taken from a tumor
in the lung contains cells that look like breast cells, the doctor
determines that the lung tumor is a secondary tumor. Still, the
determination of the primary tumor can often be very difficult, and the
pathologist may have to use several adjuvant techniques, such as immunohistochemistry, FISH (fluorescent in situ hybridization), and others. Despite the use of techniques, in some cases the primary tumor remains unidentified.
Metastatic cancers may be found at the same time as the primary
tumor, or months or years later. When a second tumor is found in a
patient that has been treated for cancer in the past, it is more often a
metastasis than another primary tumor.
It was previously thought that most cancer cells have a low
metastatic potential and that there are rare cells that develop the
ability to metastasize through the development of somatic mutations.
According to this theory, diagnosis of metastatic cancers is only
possible after the event of metastasis. Traditional means of diagnosing
cancer (e.g. a biopsy)
would only investigate a subpopulation of the cancer cells and would
very likely not sample from the subpopulation with metastatic potential.
The somatic
mutation theory of metastasis development has not been substantiated in
human cancers. Rather, it seems that the genetic state of the primary
tumor reflects the ability of that cancer to metastasize. Research comparing gene expression between primary and metastatic adenocarcinomas
identified a subset of genes whose expression could distinguish primary
tumors from metastatic tumors, dubbed a "metastatic signature." Up-regulated genes in the signature include: SNRPF, HNRPAB, DHPS and securin. Actin, myosin and MHC class II
down-regulation was also associated with the signature. Additionally,
the metastatic-associated expression of these genes was also observed in
some primary tumors, indicating that cells with the potential to
metastasize could be identified concurrently with diagnosis of the
primary tumor. Recent work identified a form of genetic instability in cancer called chromosome instability (CIN) as a driver of metastasis.
In aggressive cancer cells, loose DNA fragments from unstable
chromosomes spill in the cytosol leading to the chronic activation of
innate immune pathways, which are hijacked by cancer cells to spread to
distant organs.
Expression of this metastatic signature has been correlated with a
poor prognosis and has been shown to be consistent in several types of
cancer. Prognosis was shown to be worse for individuals whose primary
tumors expressed the metastatic signature. Additionally, the expression of these metastatic-associated genes was shown to apply to other cancer types in addition to adenocarcinoma. Metastases of breast cancer, medulloblastoma and prostate cancer all had similar expression patterns of these metastasis-associated genes.
The identification of this metastasis-associated signature
provides promise for identifying cells with metastatic potential within
the primary tumor and hope for improving the prognosis of these
metastatic-associated cancers. Additionally, identifying the genes
whose expression is changed in metastasis offers potential targets to
inhibit metastasis.
Cut surface of a humerus sawn lengthwise, showing a large cancerous metastasis (the whitish tumor between the head and the shaft of the bone)
Treatment
and survival is determined, to a great extent, by whether or not a
cancer remains localized or spreads to other locations in the body. If
the cancer metastasizes to other tissues or organs it usually
dramatically increases a patient's likelihood of death. Some
cancers—such as some forms of leukemia, a cancer of the blood, or malignancies in the brain—can kill without spreading at all.
Once a cancer has metastasized it may still be treated with radiosurgery, chemotherapy, radiation therapy, biological therapy, hormone therapy, surgery,
or a combination of these interventions ("multimodal therapy"). The
choice of treatment depends on many factors, including the type of
primary cancer,
the size and location of the metastases, the patient's age and general
health, and the types of treatments used previously. In patients
diagnosed with CUP it is often still possible to treat the disease even
when the primary tumor cannot be located.
Current treatments are rarely able to cure metastatic cancer though some tumors, such as testicular cancer and thyroid cancer, are usually curable.
Palliative care,
care aimed at improving the quality of life of people with major
illness, has been recommended as part of management programs for
metastasis.
Research
Although metastasis is widely accepted to be the result of the tumor cells migration, there is a hypothesis saying that some metastases are the result of inflammatory processes by abnormal immune cells.
The existence of metastatic cancers in the absence of primary tumors
also suggests that metastasis is not always caused by malignant cells
that leave primary tumors.
The research done by Sarna's team proved that heavily pigmented melanoma cells have Young's modulus about 4.93, when in non-pigmented ones it was only 0.98. In another experiment they found that elasticity
of melanoma cells is important for its metastasis and growth:
non-pigmented tumors were bigger than pigmented and it was much easier
for them to spread. They shown that there are both pigmented and
non-pigmented cells in melanoma tumors, so that they can both be drug-resistant and metastatic.
History
In
March 2014 researchers discovered the oldest complete example of a human
with metastatic cancer. The tumors had developed in a 3,000-year-old
skeleton found in 2013 in a tomb in Sudan
dating back to 1200 BC. The skeleton was analyzed using radiography and
a scanning electron microscope. These findings were published in the Public Library of Science journal.
Etymology
Metastasis is a Greek word meaning "displacement", from μετά, meta, "next", and στάσις, stasis, "placement".
All mammalian cells descended from a fertilized egg (a zygote)
share a common DNA sequence (except for new mutations in some
lineages). However, during development and formation of different
tissues epigenetic factors change. The changes include histone modifications, CpG island methylations and chromatin reorganizations which can cause the stable silencing or activation of particular genes.
Once differentiated tissues are formed, CpG island methylation is
generally stably inherited from one cell division to the next through
the DNA methylation maintenance machinery.
In cancer, a number of mutational changes are found in protein coding genes. Colorectal cancers typically have 3 to 6 driver mutations and 33 to 66 hitchhiker or passenger mutations that silence protein expression in the genes affected. However, transcriptional silencing may be more important than mutation in causing gene silencing in progression to cancer.
In colorectal cancers about 600 to 800 genes are transcriptionally
silenced, compared to adjacent normal-appearing tissues, by CpG island
methylation. Transcriptional repression in cancer can also occur by
other epigenetic mechanisms, such as altered expression of microRNAs.
CpG islands are frequent control elements
CpG islands are commonly 200 to 2000 base pairs long, have a C:G base pair content >50%, and have frequent 5' → 3' CpG sequences. About 70% of human promoters located near the transcription start site of a gene contain a CpG island.
Promoters located at a distance from the transcription start site of a gene also frequently contain CpG islands. The promoter of the DNA repair gene ERCC1, for instance, was identified and located about 5,400 nucleotides upstream of its coding region. CpG islands also occur frequently in promoters for functional noncoding RNAs such as microRNAs and Long non-coding RNAs (lncRNAs).
Methylation of CpG islands in promoters stably silences genes
Genes can be silenced by multiple methylation of CpG sites in the CpG islands of their promoters.
Even if silencing of a gene is initiated by another mechanism, this
often is followed by methylation of CpG sites in the promoter CpG island
to stabilize the silencing of the gene. On the other hand, hypomethylation of CpG islands in promoters can result in gene over-expression.
Promoter CpG hyper/hypo-methylation in cancer
In
cancers, loss of expression of genes occurs about 10 times more
frequently by hypermethylation of promoter CpG islands than by
mutations. For instance, in colon tumors compared to adjacent
normal-appearing colonic mucosa, about 600 to 800 heavily methylated CpG
islands occur in promoters of genes in the tumors while these CpG
islands are not methylated in the adjacent mucosa. In contrast, as Vogelstein et al. point out, in a colorectal cancer there are typically only about 3 to 6 driver mutations and 33 to 66 hitchhiker or passenger mutations.
DNA repair gene silencing in cancer
In
sporadic cancers, a DNA repair deficiency is occasionally found to be
due to a mutation in a DNA repair gene. However, much more frequently,
reduced or absent expression of a DNA repair gene in cancer is due to
methylation of its promoter. For example, of 113 colorectal cancers
examined, only four had a missense mutation in the DNA repair gene MGMT, while the majority had reduced MGMT expression due to methylation of the MGMT promoter region. Similarly, among 119 cases of mismatch repair-deficient colorectal cancers that lacked DNA repair gene PMS2 expression, 6 had a mutation in the PMS2
gene, while for 103 PMS2 was deficient because its pairing partner MLH1
was repressed due to promoter methylation (PMS2 protein is unstable in
the absence of MLH1).
In the remaining 10 cases, loss of PMS2 expression was likely due to
epigenetic overexpression of the microRNA, miR-155, which down-regulates
MLH1.
Frequency of hypermethylation of DNA repair genes in cancer
Twenty-two
DNA repair genes with hypermethylated promoters, and reduced or absent
expression, were found to occur among 17 types of cancer, as listed in
two review articles. Promoter hypermethylation of MGMT
occurs frequently in a number of cancers including 93% of bladder
cancers, 88% of stomach cancers, 74% of thyroid cancers, 40%-90% of
colorectal cancers and 50% of brain cancers. That review also indicated promoter hypermethylation of LIG4, NEIL1, ATM, MLH1 or FANCB occurs at frequencies between 33% to 82% in one or more of head and neck cancers, non-small-cell lung cancers or non-small-cell lung cancer
squamous cell carcinomas. The article [Epigenetic inactivation of the
premature aging Werner syndrome gene in human cancer] indicates the DNA
repair gene WRN has a promoter that is frequently hypermethylated in a number of cancers, with hypermethylation occurring in 11% to 38% of colorectal, head and neck, stomach, prostate, breast, thyroid, non-Hodgkin lymphoma, chondrosarcoma and osteosarcoma cancers (see WRN).
Likely role of hypermethylation of DNA repair genes in cancer
As discussed by Jin and Roberston in their review,
silencing of a DNA repair gene by hypermethylation may be a very early
step in progression to cancer. Such silencing is proposed to act
similarly to a germ-line mutation in a DNA repair gene, and predisposes
the cell and its descendants to progression to cancer. Another review
also indicated an early role for hypermethylation of DNA repair genes
in cancer. If a gene necessary for DNA repair is hypermethylated,
resulting in deficient DNA repair, DNA damages will accumulate.
Increased DNA damage tends to cause increased errors during DNA
synthesis, leading to mutations that can give rise to cancer.
If hypermethylation of a DNA repair gene is an early step in
carcinogenesis, then it may also occur in the normal-appearing tissues
surrounding the cancer from which the cancer arose (the field defect). See the table below.
Frequencies of hypermethylated promoters in DNA repair genes in sporadic cancers and in adjacent field defects
Cancer
Gene
Frequency in Cancer
Frequency in Field Defect
Colorectal
MGMT
55%
54%
Colorectal
MSH2
13%
5%
Colorectal
WRN
29%
13%
Head and Neck
MGMT
54%
38%
Head and Neck
MLH1
33%
25%
Non-small cell lung cancer
ATM
69%
59%
Non-small cell lung cancer
MLH1
69%
72%
omach
MGMT
88%
78%
Stomach
MLH1
73%
20%
Esophagus
MLH1
77%-100%
23%-79%
While DNA damages may give rise to mutations through error prone translesion synthesis, DNA damages can also give rise to epigenetic alterations during faulty DNA repair processes.
The DNA damages that accumulate due to hypermethylation of the
promoters of DNA repair genes can be a source of the increased
epigenetic alterations found in many genes in cancers.
In an early study, looking at a limited set of transcriptional promoters, Fernandez et al.
examined the DNA methylation profiles of 855 primary tumors. Comparing
each tumor type with its corresponding normal tissue, 729 CpG island
sites (55% of the 1322 CpG island sites evaluated) showed differential
DNA methylation. Of these sites, 496 were hypermethylated (repressed)
and 233 were hypomethylated (activated). Thus, there is a high level of
promoter methylation alterations in tumors. Some of these alterations
may contribute to cancer progression.
DNA methylation of microRNAs in cancer
In mammals, microRNAs (miRNAs) regulate the transcriptional activity of about 60% of protein-encoding genes.
Individual miRNAs can each target, and repress transcription of, on
average, roughly 200 messenger RNAs of protein coding genes. The promoters of about one third of the 167 miRNAs evaluated by Vrba et al.
in normal breast tissues were differentially hyper/hypo-methylated in
breast cancers. A more recent study pointed out that the 167 miRNAs
evaluated by Vrba et al. were only 10% of the miRNAs found expressed in
breast tissues. This later study found that 58% of the miRNAs in breast tissue had differentially methylated regions in their promoters in breast cancers, including 278 hypermethylated miRNAs and 802 hypomethylated miRNAs.
One miRNA that is over-expressed about 100-fold in breast cancers is miR-182. MiR-182 targets the BRCA1 messenger RNA and may be a major cause of reduced BRCA1 protein expression in many breast cancers.
microRNAs that control DNA methyltransferase genes in cancer
Some miRNAs target the messenger RNAs for DNA methyltransferase genes DNMT1, DNMT3A and DNMT3B, whose gene products are needed for initiating and stabilizing promoter methylations. As summarized in three reviews,
miRNAs miR-29a, miR-29b and miR-29c target DNMT3A and DNMT3B; miR-148a
and miR-148b target DNMT3B; and miR-152 and miR-301 target DNMT1. In
addition, miR-34b targets DNMT1 and the promoter of miR-34b itself is
hypermethylated and under-expressed in the majority of prostate cancers.
When expression of these microRNAs is altered, they may also be a
source of the hyper/hypo-methylation of the promoters of protein-coding
genes in cancers.
Representation of a DNA molecule that is methylated. The two white spheres represent methyl groups. They are bound to two cytosinenucleotide molecules that make up the DNA sequence.
DNA methylation is a biological process by which methyl groups are added to the DNA molecule. Methylation can change the activity of a DNA segment without changing the sequence. When located in a gene promoter, DNA methylation typically acts to repress gene transcription. In mammals, DNA methylation is essential for normal development and is associated with a number of key processes including genomic imprinting, X-chromosome inactivation, repression of transposable elements, aging, and carcinogenesis.
As of 2016, two nucleobases have been found on which natural, enzymatic DNA methylation takes place: adenine and cytosine. The modified bases are N6-methyladenine, 5-methylcytosine and N4-methylcytosine.
Methylation of cytosine to form 5-methylcytosine occurs at the same 5 position on the pyrimidine ring where the DNA base thymine's methyl group is located; the same position distinguishes thymine from the analogous RNA base uracil, which has no methyl group. Spontaneous deamination of 5-methylcytosine
converts it to thymine. This results in a T:G mismatch. Repair
mechanisms then correct it back to the original C:G pair; alternatively,
they may substitute A for G, turning the original C:G pair into a T:A
pair, effectively changing a base and introducing a mutation. This
misincorporated base will not be corrected during DNA replication as
thymine is a DNA base. If the mismatch is not repaired and the cell
enters the cell cycle the strand carrying the T will be complemented by
an A in one of the daughter cells, such that the mutation becomes
permanent. The near-universal use of thymine exclusively in DNA and uracil
exclusively in RNA may have evolved as an error-control mechanism, to
facilitate the removal of uracils generated by the spontaneous
deamination of cytosine.
DNA methylation as well as many of its contemporary DNA
methyltransferases have been thought to evolve from early world
primitive RNA methylation activity and is supported by several lines of
evidence.
In plants and other organisms, DNA methylation is found in three different sequence contexts: CG (or CpG),
CHG or CHH (where H correspond to A, T or C). In mammals however, DNA
methylation is almost exclusively found in CpG dinucleotides, with the
cytosines on both strands being usually methylated. Non-CpG methylation
can however be observed in embryonic stem cells, and has also been indicated in neural development. Furthermore, non-CpG methylation has also been observed in hematopoietic progenitor cells, and it occurred mainly in a CpApC sequence context.
Conserved function of DNA methylation
Typical DNA methylation landscape in mammals
The DNA methylation landscape of vertebrates is very particular
compared to other organisms. In mammals, around 75% of CpG dinucleotides
are methylated in somatic cells, and DNA methylation appears as a default state that has to be specifically excluded from defined locations.
By contrast, the genome of most plants, invertebrates, fungi, or
protists show “mosaic” methylation patterns, where only specific genomic
elements are targeted, and they are characterized by the alternation of
methylated and unmethylated domains.
High CpG methylation in mammalian genomes has an evolutionary
cost because it increases the frequency of spontaneous mutations. Loss
of amino-groups occurs with a high frequency for cytosines, with
different consequences depending on their methylation. Methylated C
residues spontaneously deaminate to form T residues over time; hence CpG
dinucleotides steadily deaminate to TpG dinucleotides, which is
evidenced by the under-representation of CpG dinucleotides in the human
genome (they occur at only 21% of the expected frequency).
(On the other hand, spontaneous deamination of unmethylated C residues
gives rise to U residues, a change that is quickly recognized and
repaired by the cell.)
CpG islands
In
mammals, the only exception for this global CpG depletion resides in a
specific category of GC- and CpG-rich sequences termed CpG islands that
are generally unmethylated and therefore retained the expected CpG
content.
CpG islands are usually defined as regions with: 1) a length greater
than 200bp, 2) a G+C content greater than 50%, 3) a ratio of observed to
expected CpG greater than 0.6, although other definitions are sometimes
used. Excluding repeated sequences, there are around 25,000 CpG islands in the human genome, 75% of which being less than 850bp long.
They are major regulatory units and around 50% of CpG islands are
located in gene promoter regions, while another 25% lie in gene bodies,
often serving as alternative promoters. Reciprocally, around 60-70% of
human genes have a CpG island in their promoter region. The majority of CpG islands are constitutively unmethylated and enriched for permissive chromatin modification
such as H3K4 methylation. In somatic tissues, only 10% of CpG islands
are methylated, the majority of them being located in intergenic and
intragenic regions.
Repression of CpG-dense promoters
DNA
methylation was probably present at some extent in very early eukaryote
ancestors. In virtually every organism analyzed, methylation in
promoter regions correlates negatively with gene expression.
CpG-dense promoters of actively transcribed genes are never methylated,
but, reciprocally, transcriptionally silent genes do not necessarily
carry a methylated promoter. In mouse and human, around 60–70% of genes
have a CpG island in their promoter region and most of these CpG islands
remain unmethylated independently of the transcriptional activity of
the gene, in both differentiated and undifferentiated cell types.
Of note, whereas DNA methylation of CpG islands is unambiguously linked
with transcriptional repression, the function of DNA methylation in
CG-poor promoters remains unclear; albeit there is little evidence that
it could be functionally relevant.
DNA methylation may affect the transcription of genes in two
ways. First, the methylation of DNA itself may physically impede the
binding of transcriptional proteins to the gene, and second, and likely more important, methylated DNA may be bound by proteins known as methyl-CpG-binding domain proteins (MBDs). MBD proteins then recruit additional proteins to the locus, such as histone deacetylases and other chromatin remodeling proteins that can modify histones, thereby forming compact, inactive chromatin, termed heterochromatin. This link between DNA methylation and chromatin structure is very important. In particular, loss of methyl-CpG-binding protein 2 (MeCP2) has been implicated in Rett syndrome; and methyl-CpG-binding domain protein 2 (MBD2) mediates the transcriptional silencing of hypermethylated genes in "cancer".
Repression of transposable elements
DNA
methylation is a powerful transcriptional repressor, at least in CpG
dense contexts. Transcriptional repression of protein-coding genes
appears essentially limited to very specific classes of genes that need
to be silent permanently and in almost all tissues. While DNA
methylation does not have the flexibility required for the fine-tuning
of gene regulation, its stability is perfect to ensure the permanent
silencing of transposable elements.
Transposon control is one of the most ancient functions of DNA
methylation that is shared by animals, plants and multiple protists. It is even suggested that DNA methylation evolved precisely for this purpose.
Genome expansion
DNA
methylation of transposable elements has been known to be related to
genome expansion. However, the evolutionary driver for genome expansion
remains unknown. There is a clear correlation between the size of the
genome and CpG, suggesting that the DNA methylation of transposable
elements led to a noticeable increase in the mass of DNA.
Methylation of the gene body of highly transcribed genes
A
function that appears even more conserved than transposon silencing is
positively correlated with gene expression. In almost all species where
DNA methylation is present, DNA methylation is especially enriched in
the body of highly transcribed genes. The function of gene body methylation is not well understood. A body of evidence suggests that it could regulate splicing and suppress the activity of intragenic transcriptional units (cryptic promoters or transposable elements).
Gene-body methylation appears closely tied to H3K36 methylation. In
yeast and mammals, H3K36 methylation is highly enriched in the body of
highly transcribed genes. In yeast at least, H3K36me3 recruits enzymes such as histone deacetylases to condense chromatin and prevent the activation of cryptic start sites.
In mammals, DNMT3a and DNMT3b PWWP domain binds to H3K36me3 and the two
enzymes are recruited to the body of actively transcribed genes.
In mammals
Dynamic
of DNA methylation during mouse embryonic development. E3.5-E6, etc.,
refer to days after fertilization. PGC: primordial germ cells
DNA methylation patterns are largely erased and then re-established
between generations in mammals. Almost all of the methylations from the
parents are erased, first during gametogenesis, and again in early embryogenesis,
with demethylation and remethylation occurring each time. Demethylation
in early embryogenesis occurs in the preimplantation period in two
stages – initially in the zygote, then during the first few embryonic replication cycles of morula and blastula.
A wave of methylation then takes place during the implantation stage of
the embryo, with CpG islands protected from methylation. This results
in global repression and allows housekeeping genes to be expressed in
all cells. In the post-implantation stage, methylation patterns are
stage- and tissue-specific, with changes that would define each
individual cell type lasting stably over a long period.
Whereas DNA methylation is not necessary per se for
transcriptional silencing, it is thought nonetheless to represent a
“locked” state that definitely inactivates transcription. In particular,
DNA methylation appears critical for the maintenance of mono-allelic
silencing in the context of genomic imprinting and X chromosome inactivation.
In these cases, expressed and silent alleles differ by their
methylation status, and loss of DNA methylation results in loss of
imprinting and re-expression of Xist in somatic cells. During embryonic
development, few genes change their methylation status, at the important
exception of many genes specifically expressed in the germline. DNA methylation appears absolutely required in differentiated cells,
as knockout of any of the three competent DNA methyltransferase results
in embryonic or post-partum lethality. By contrast, DNA methylation is
dispensable in undifferentiated cell types, such as the inner cell mass
of the blastocyst, primordial germ cells or embryonic stem cells. Since
DNA methylation appears to directly regulate only a limited number of
genes, how precisely DNA methylation absence causes the death of
differentiated cells remain an open question.
Due to the phenomenon of genomic imprinting, maternal and paternal genomes are differentially marked and must be properly reprogrammed every time they pass through the germline. Therefore, during gametogenesis,
primordial germ cells must have their original biparental DNA
methylation patterns erased and re-established based on the sex of the
transmitting parent. After fertilization, the paternal and maternal
genomes are once again demethylated and remethylated (except for
differentially methylated regions associated with imprinted genes). This
reprogramming is likely required for totipotency of the newly formed
embryo and erasure of acquired epigenetic changes.
In many disease processes, such as cancer, gene promoter CpG islands acquire abnormal hypermethylation, which results in transcriptional silencing that can be inherited by daughter cells following cell division.
Alterations of DNA methylation have been recognized as an important
component of cancer development. Hypomethylation, in general, arises
earlier and is linked to chromosomal instability and loss of imprinting,
whereas hypermethylation is associated with promoters and can arise
secondary to gene (oncogene suppressor) silencing, but might be a target
for epigenetic therapy.
Global hypomethylation has also been implicated in the development and progression of cancer through different mechanisms. Typically, there is hypermethylation of tumor suppressor genes and hypomethylation of oncogenes.
Generally, in progression to cancer, hundreds of genes are silenced or activated.
Although silencing of some genes in cancers occurs by mutation, a large
proportion of carcinogenic gene silencing is a result of altered DNA
methylation (see DNA methylation in cancer). DNA methylation causing silencing in cancer typically occurs at multiple CpG sites in the CpG islands that are present in the promoters of protein coding genes.
Silencing of DNA repair genes through methylation of CpG islands
in their promoters appears to be especially important in progression to
cancer (see methylation of DNA repair genes in cancer).
In atherosclerosis
Epigenetic modifications such as DNA methylation have been implicated in cardiovascular disease, including atherosclerosis.
In animal models of atherosclerosis, vascular tissue, as well as blood
cells such as mononuclear blood cells, exhibit global hypomethylation
with gene-specific areas of hypermethylation. DNA methylation
polymorphisms may be used as an early biomarker of atherosclerosis since
they are present before lesions are observed, which may provide an
early tool for detection and risk prevention.
Two of the cell types targeted for DNA methylation polymorphisms
are monocytes and lymphocytes, which experience an overall
hypomethylation. One proposed mechanism behind this global
hypomethylation is elevated homocysteine levels causing hyperhomocysteinemia,
a known risk factor for cardiovascular disease. High plasma levels of
homocysteine inhibit DNA methyltransferases, which causes
hypomethylation. Hypomethylation of DNA affects genes that alter smooth
muscle cell proliferation, cause endothelial cell dysfunction, and
increase inflammatory mediators, all of which are critical in forming
atherosclerotic lesions. High levels of homocysteine also result in hypermethylation of CpG islands in the promoter region of the estrogen receptor alpha (ERα) gene, causing its down regulation.
ERα protects against atherosclerosis due to its action as a growth
suppressor, causing the smooth muscle cells to remain in a quiescent
state.
Hypermethylation of the ERα promoter thus allows intimal smooth muscle
cells to proliferate excessively and contribute to the development of
the atherosclerotic lesion.
Another gene that experiences a change in methylation status in atherosclerosis is the monocarboxylate transporter
(MCT3), which produces a protein responsible for the transport of
lactate and other ketone bodies out of many cell types, including
vascular smooth muscle cells. In atherosclerosis patients, there is an
increase in methylation of the CpG islands in exon 2, which decreases
MCT3 protein expression. The downregulation of MCT3 impairs lactate
transport and significantly increases smooth muscle cell proliferation,
which further contributes to the atherosclerotic lesion. An ex vivo
experiment using the demethylating agent Decitabine
(5-aza-2 -deoxycytidine) was shown to induce MCT3 expression in a dose
dependent manner, as all hypermethylated sites in the exon 2 CpG island
became demethylated after treatment. This may serve as a novel
therapeutic agent to treat atherosclerosis, although no human studies
have been conducted thus far.
In heart failure
In addition to atherosclerosis
described above, specific epigenetic changes have been identified in
the failing human heart. This may vary by disease etiology. For example,
in ischemic heart failure DNA methylation changes have been linked to
changes in gene expression that may direct gene expression associated
with the changes in heart metabolism known to occur.
Additional forms of heart failure (e.g. diabetic cardiomyopathy) and
co-morbidities (e.g. obesity) must be explored to see how common these
mechanisms are. Most strikingly, in failing human heart these changes in
DNA methylation are associated with racial and socioeconomic status
which further impact how gene expression is altered, and may influence how the individual's heart failure should be treated.
In aging
In
humans and other mammals, DNA methylation levels can be used to
accurately estimate the age of tissues and cell types, forming an
accurate epigenetic clock.
A longitudinal study of twin
children showed that, between the ages of 5 and 10, there was
divergence of methylation patterns due to environmental rather than
genetic influences. There is a global loss of DNA methylation during aging.
In a study that analyzed the complete DNA methylomes of CD4+T cells
in a newborn, a 26 years old individual and a 103 years old individual
were observed that the loss of methylation is proportional to age.
Hypomethylated CpGs observed in the centenarian DNAs compared with the
neonates covered all genomic compartments (promoters, intergenic,
intronic and exonic regions). However, some genes become hypermethylated with age, including genes for the estrogen receptor, p16, and insulin-like growth factor 2.
In exercise
High intensity exercise has been shown to result in reduced DNA methylation in skeletal muscle. Promoter methylation of PGC-1α and PDK4 were immediately reduced after high intensity exercise, whereas PPAR-γ methylation was not reduced until three hours after exercise. At the same time, six months of exercise in previously sedentary middle-age men resulted in increased methylation in adipose tissue. One study showed a possible increase in global genomic DNA methylation of white blood cells with more physical activity in non-Hispanics.
In B-cell differentiation
A study that investigated the methylome of B cells along their differentiation cycle, using whole-genome bisulfite sequencing
(WGBS), showed that there is a hypomethylation from the earliest stages
to the most differentiated stages. The largest methylation difference
is between the stages of germinal center B cells and memory B cells.
Furthermore, this study showed that there is a similarity between B cell
tumors and long-lived B cells in their DNA methylation signatures.
In the brain
Two reviews summarize evidence that DNA methylation alterations in brain neurons are important in learning and memory. Contextual fear conditioning (a form of associative learning) in animals, such as mice and rats, is rapid and is extremely robust in creating memories. In mice and in rats
contextual fear conditioning, within 1–24 hours, it is associated with
altered methylations of several thousand DNA cytosines in genes of hippocampus neurons. Twenty four hours after contextual fear conditioning, 9.2% of the genes in rat hippocampus neurons are differentially methylated. In mice,
when examined at four weeks after conditioning, the hippocampus
methylations and demethylations had been reset to the original naive
conditions. The hippocampus
is needed to form memories, but memories are not stored there. For such
mice, at four weeks after contextual fear conditioning, substantial
differential CpG methylations and demethylations occurred in cortical neurons during memory maintenance, and there were 1,223 differentially methylated genes in their anterior cingulate cortex. Active changes in neuronal DNA methylation and demethylation appear to act as controllers of synaptic scaling and glutamate receptor trafficking in learning and memory formation.
In mammalian cells, DNA methylation occurs mainly at the C5 position
of CpG dinucleotides and is carried out by two general classes of
enzymatic activities – maintenance methylation and de novo methylation.
Maintenance methylation activity is necessary to preserve DNA
methylation after every cellular DNA replication cycle. Without the DNA methyltransferase
(DNMT), the replication machinery itself would produce daughter strands
that are unmethylated and, over time, would lead to passive
demethylation. DNMT1 is the proposed maintenance methyltransferase that
is responsible for copying DNA methylation patterns to the daughter
strands during DNA replication. Mouse models with both copies of DNMT1
deleted are embryonic lethal at approximately day 9, due to the
requirement of DNMT1 activity for development in mammalian cells.
It is thought that DNMT3a and DNMT3b are the de novo
methyltransferases that set up DNA methylation patterns early in
development. DNMT3L is a protein that is homologous to the other DNMT3s
but has no catalytic activity. Instead, DNMT3L assists the de novo
methyltransferases by increasing their ability to bind to DNA and
stimulating their activity. Mice and rats have a third functional de novo methyltransferase enzyme named DNMT3C, which evolved as a paralog of Dnmt3b
by tandem duplication in the common ancestral of Muroidea rodents.
DNMT3C catalyzes the methylation of promoters of transposable elements
during early spermatogenesis, an activity shown to be essential for
their epigenetic repression and male fertility. It is yet unclear if in other mammals that do not have DNMT3C (like humans) rely on DNMT3B or DNMT3A for de novo methylation of transposable elements in the germline. Finally, DNMT2 (TRDMT1)
has been identified as a DNA methyltransferase homolog, containing all
10 sequence motifs common to all DNA methyltransferases; however, DNMT2
(TRDMT1) does not methylate DNA but instead methylates cytosine-38 in
the anticodon loop of aspartic acid transfer RNA.
Since many tumor suppressor genes are silenced by DNA methylation during carcinogenesis, there have been attempts to re-express these genes by inhibiting the DNMTs. 5-Aza-2'-deoxycytidine (decitabine) is a nucleoside analog
that inhibits DNMTs by trapping them in a covalent complex on DNA by
preventing the β-elimination step of catalysis, thus resulting in the
enzymes' degradation. However, for decitabine to be active, it must be
incorporated into the genome
of the cell, which can cause mutations in the daughter cells if the
cell does not die. In addition, decitabine is toxic to the bone marrow,
which limits the size of its therapeutic window. These pitfalls have led
to the development of antisense RNA therapies that target the DNMTs by
degrading their mRNAs and preventing their translation.
However, it is currently unclear whether targeting DNMT1 alone is
sufficient to reactivate tumor suppressor genes silenced by DNA
methylation.
In plants
Significant progress has been made in understanding DNA methylation in the model plant Arabidopsis thaliana.
DNA methylation in plants differs from that of mammals: while DNA
methylation in mammals mainly occurs on the cytosine nucleotide in a CpG site,
in plants the cytosine can be methylated at CpG, CpHpG, and CpHpH
sites, where H represents any nucleotide but not guanine. Overall, Arabidopsis DNA is highly methylated, mass spectrometry analysis estimated 14% of cytosines to be modified.
The principal Arabidopsis DNA methyltransferase enzymes,
which transfer and covalently attach methyl groups onto DNA, are DRM2,
MET1, and CMT3. Both the DRM2 and MET1 proteins share significant
homology to the mammalian methyltransferases DNMT3 and DNMT1,
respectively, whereas the CMT3 protein is unique to the plant kingdom.
There are currently two classes of DNA methyltransferases: 1) the de novo
class or enzymes that create new methylation marks on the DNA; 2) a
maintenance class that recognizes the methylation marks on the parental
strand of DNA and transfers new methylation to the daughter strands
after DNA replication. DRM2 is the only enzyme that has been implicated
as a de novo DNA methyltransferase. DRM2 has also been shown,
along with MET1 and CMT3 to be involved in maintaining methylation marks
through DNA replication. Other DNA methyltransferases are expressed in plants but have no known function (see the Chromatin Database).
It is not clear how the cell determines the locations of de novo DNA methylation, but evidence suggests that for many (though not all) locations, RNA-directed DNA methylation
(RdDM) is involved. In RdDM, specific RNA transcripts are produced from
a genomic DNA template, and this RNA forms secondary structures called
double-stranded RNA molecules. The double-stranded RNAs, through either the small interfering RNA (siRNA) or microRNA (miRNA) pathways direct de-novo DNA methylation of the original genomic location that produced the RNA. This sort of mechanism is thought to be important in cellular defense against RNA viruses and/or transposons,
both of which often form a double-stranded RNA that can be mutagenic to
the host genome. By methylating their genomic locations, through an as
yet poorly understood mechanism, they are shut off and are no longer
active in the cell, protecting the genome from their mutagenic effect.
Recently, it was described that methylation of the DNA is the main
determinant of embryogenic cultures formation from explants in woody
plants and is regarded the main mechanism that explains the poor
response of mature explants to somatic embryogenesis in the plants (Isah
2016).
Diverse orders of insects show varied patterns of DNA methylation, from almost undetectable levels in flies to low levels in butterflies and higher in true bugs and some cockroaches (up to 14% of all CG sites in Blattella asahinai).
Functional DNA methylation has been discovered in Honey Bees.
DNA methylation marks are mainly on the gene body, and current opinions
on the function of DNA methylation is gene regulation via alternative
splicing
DNA methylation levels in Drosophila melanogaster are nearly undetectable. Sensitive methods applied to Drosophila DNA Suggest levels in the range of 0.1–0.3% of total cytosine. This low level of methylation
appears to reside in genomic sequence patterns that are very different
from patterns seen in humans, or in other animal or plant species to
date. Genomic methylation in D. melanogaster was found at specific short
motifs (concentrated in specific 5-base sequence motifs that are CA-
and CT-rich but depleted of guanine) and is independent of DNMT2
activity. Further, highly sensitive mass spectrometry approaches,
have now demonstrated the presence of low (0.07%) but significant
levels of adenine methylation during the earliest stages of Drosophila
embryogenesis.
In fungi
Many fungi have low levels (0.1 to 0.5%) of cytosine methylation, whereas other fungi have as much as 5% of the genome methylated. This value seems to vary both among species and among isolates of the same species. There is also evidence that DNA methylation may be involved in state-specific control of gene expression in fungi. However, at a detection limit of 250 attomoles by using ultra-high sensitive mass spectrometry DNA methylation was not confirmed in single cellular yeast species such as Saccharomyces cerevisiae or Schizosaccharomyces pombe, indicating that yeasts do not possess this DNA modification.
Although brewers' yeast (Saccharomyces), fission yeast (Schizosaccharomyces), and Aspergillus flavus have no detectable DNA methylation, the model filamentous fungus Neurospora crassa has a well-characterized methylation system. Several genes control methylation in Neurospora and mutation of the DNA methyl transferase, dim-2, eliminates all DNA methylation but does not affect growth or sexual reproduction. While the Neurospora genome has very little repeated DNA, half of the methylation occurs in repeated DNA including transposon
relics and centromeric DNA. The ability to evaluate other important
phenomena in a DNA methylase-deficient genetic background makes Neurospora an important system in which to study DNA methylation.
In other eukaryotes
DNA methylation is largely absent from Dictyostelium discoidium where it appears to occur at about 0.006% of cytosines. In contrast, DNA methylation is widely distributed in Physarum polycephalum where 5-methylcytosine makes up as much as 8% of total cytosine.
In bacteria
All methylations in a prokaryote.
In some prokaryotic organisms, all three previously known DNA
methylation types are represented (N4-methylcytosine: m4C,
5-methylcytosine: m5C and N6-methyladenine: m6A). Six examples are shown
here, two of which belong to the Archaea domain and four of which
belong to the Bacteria domain. The information comes from Blow et al.
(2016).
In the left column are the species names of the organisms, to the right
there are examples of methylated DNA motifs. The full names of the
archaea and bacterial strains are according to the NCBI taxonomy:
"Methanocaldococcus jannaschii DSM 2661", "Methanocorpusculum labreanum
Z", "Clostridium perfringens ATCC 13127", "Geopsychrobacter
electrodiphilus DSM 16401", "Rhodopseudomonas palustris CGA009" and
"Salmonella enterica subsp. enterica serovar Paratyphi A str. ATCC 9150 "
Adenine or cytosine methylation are mediated by restriction modification systems of many bacteria, in which specific DNA sequences are methylated periodically throughout the genome. A methylase
is the enzyme that recognizes a specific sequence and methylates one of
the bases in or near that sequence. Foreign DNAs (which are not
methylated in this manner) that are introduced into the cell are
degraded by sequence-specific restriction enzymes
and cleaved. Bacterial genomic DNA is not recognized by these
restriction enzymes. The methylation of native DNA acts as a sort of
primitive immune system, allowing the bacteria to protect themselves
from infection by bacteriophage.
E. coliDNA adenine methyltransferase (Dam) is an enzyme of ~32 kDa that does not belong to a restriction/modification system. The target recognition sequence for E. coli
Dam is GATC, as the methylation occurs at the N6 position of the
adenine in this sequence (G meATC). The three base pairs flanking each
side of this site also influence DNA–Dam binding. Dam plays several key
roles in bacterial processes, including mismatch repair, the timing of
DNA replication, and gene expression. As a result of DNA replication,
the status of GATC sites in the E. coli genome changes from fully
methylated to hemimethylated. This is because adenine introduced into
the new DNA strand is unmethylated. Re-methylation occurs within two to
four seconds, during which time replication errors in the new strand are
repaired. Methylation, or its absence, is the marker that allows the
repair apparatus of the cell to differentiate between the template and
nascent strands. It has been shown that altering Dam activity in
bacteria results in an increased spontaneous mutation rate. Bacterial
viability is compromised in dam mutants that also lack certain other DNA
repair enzymes, providing further evidence for the role of Dam in DNA
repair.
One region of the DNA that keeps its hemimethylated status for longer is the origin of replication,
which has an abundance of GATC sites. This is central to the bacterial
mechanism for timing DNA replication. SeqA binds to the origin of
replication, sequestering it and thus preventing methylation. Because
hemimethylated origins of replication are inactive, this mechanism
limits DNA replication to once per cell cycle.
Expression of certain genes, for example, those coding for pilus expression in E. coli,
is regulated by the methylation of GATC sites in the promoter region of
the gene operon. The cells' environmental conditions just after DNA
replication determine whether Dam is blocked from methylating a region
proximal to or distal from the promoter region. Once the pattern of
methylation has been created, the pilus gene transcription is locked in
the on or off position until the DNA is again replicated. In E. coli, these pili operons have important roles in virulence in urinary tract infections. It has been proposed that inhibitors of Dam may function as antibiotics.
On the other hand, DNA cytosine methylase targets CCAGG and CCTGG
sites to methylate cytosine at the C5 position (C meC(A/T) GG). The
other methylase enzyme, EcoKI, causes methylation of adenines in the
sequences AAC(N6)GTGC and GCAC(N6)GTT.
In Clostridioides difficile, DNA methylation at the target motif CAAAAA was shown to impact sporulation, a key step in disease transmission, as well as cell length, biofilm formation and host colonization.
Molecular cloning
Most strains used by molecular biologists are derivatives of E. coli
K-12, and possess both Dam and Dcm, but there are commercially
available strains that are dam-/dcm- (lack of activity of either
methylase). In fact, it is possible to unmethylate the DNA extracted
from dam+/dcm+ strains by transforming it into dam-/dcm- strains. This
would help digest sequences that are not being recognized by
methylation-sensitive restriction enzymes.
The restriction enzyme
DpnI can recognize 5'-GmeATC-3' sites and digest the methylated DNA.
Being such a short motif, it occurs frequently in sequences by chance,
and as such its primary use for researchers is to degrade template DNA
following PCRs
(PCR products lack methylation, as no methylases are present in the
reaction). Similarly, some commercially available restriction enzymes
are sensitive to methylation at their cognate restriction sites and must
as mentioned previously be used on DNA passed through a dam-/dcm-
strain to allow cutting.
Detection
DNA methylation can be detected by the following assays currently used in scientific research:
Mass spectrometry
is a very sensitive and reliable analytical method to detect DNA
methylation. MS, in general, is however not informative about the
sequence context of the methylation, thus limited in studying the
function of this DNA modification.
Methylation-Specific PCR (MSP),
which is based on a chemical reaction of sodium bisulfite with DNA that
converts unmethylated cytosines of CpG dinucleotides to uracil or UpG,
followed by traditional PCR.
However, methylated cytosines will not be converted in this process,
and primers are designed to overlap the CpG site of interest, which
allows one to determine methylation status as methylated or
unmethylated.
Whole genome bisulfite sequencing,
also known as BS-Seq, which is a high-throughput genome-wide analysis
of DNA methylation. It is based on the aforementioned sodium bisulfite
conversion of genomic DNA, which is then sequenced on a Next-generation sequencing platform.
The sequences obtained are then re-aligned to the reference genome to
determine the methylation status of CpG dinucleotides based on
mismatches resulting from the conversion of unmethylated cytosines into
uracil.
Reduced representation bisulfite sequencing,
also known as RRBS knows several working protocols. The first RRBS
protocol was called RRBS and aims for around 10% of the methylome, a
reference genome is needed. Later came more protocols that were able to
sequence a smaller portion of the genome and higher sample multiplexing.
EpiGBS was the first protocol where you could multiplex 96 samples in
one lane of Illumina sequencing and were a reference genome was no
longer needed. A de novo reference construction from the Watson and
Crick reads made population screening of SNP's and SMP's simultaneously a
fact.
The HELP assay, which is based on restriction enzymes' differential ability to recognize and cleave methylated and unmethylated CpG DNA sites.
GLAD-PCR assay,
which is based on a new type of enzymes – site-specific methyl-directed
DNA endonucleases, which hydrolyze only methylated DNA.
ChIP-on-chip
assays, which is based on the ability of commercially prepared
antibodies to bind to DNA methylation-associated proteins like MeCP2.
Restriction landmark genomic scanning,
a complicated and now rarely used assay based upon restriction enzymes'
differential recognition of methylated and unmethylated CpG sites; the
assay is similar in concept to the HELP assay.
Pyrosequencing
of bisulfite treated DNA. This is the sequencing of an amplicon made by
a normal forward primer but a biotinylated reverse primer to PCR the
gene of choice. The Pyrosequencer then analyses the sample by denaturing
the DNA and adding one nucleotide at a time to the mix according to a
sequence given by the user. If there is a mismatch, it is recorded and
the percentage of DNA for which the mismatch is present is noted. This
gives the user a percentage of methylation per CpG island.
Molecular break light assay for DNA adenine methyltransferase
activity – an assay that relies on the specificity of the restriction
enzyme DpnI for fully methylated (adenine methylation) GATC sites in an
oligonucleotide labeled with a fluorophore and quencher. The adenine
methyltransferase methylates the oligonucleotide making it a substrate
for DpnI. Cutting of the oligonucleotide by DpnI gives rise to a
fluorescence increase.
Methyl Sensitive Southern Blotting is similar to the HELP assay,
although uses Southern blotting techniques to probe gene-specific
differences in methylation using restriction digests. This technique is
used to evaluate local methylation near the binding site for the probe.
MethylCpG Binding Proteins (MBPs) and fusion proteins containing
just the Methyl Binding Domain (MBD) are used to separate native DNA
into methylated and unmethylated fractions. The percentage methylation
of individual CpG islands can be determined by quantifying the amount of
the target in each fraction. Extremely sensitive detection can be achieved in FFPE tissues with abscription-based detection.
High Resolution Melt Analysis (HRM or HRMA), is a post-PCR
analytical technique. The target DNA is treated with sodium bisulfite,
which chemically converts unmethylated cytosines into uracils, while
methylated cytosines are preserved. PCR amplification is then carried
out with primers designed to amplify both methylated and unmethylated
templates. After this amplification, highly methylated DNA sequences
contain a higher number of CpG sites compared to unmethylated templates,
which results in a different melting temperature that can be used in
quantitative methylation detection.
Ancient DNA methylation reconstruction, a method to reconstruct
high-resolution DNA methylation from ancient DNA samples. The method is
based on the natural degradation processes that occur in ancient DNA:
with time, methylated cytosines are degraded into thymines, whereas
unmethylated cytosines are degraded into uracils. This asymmetry in
degradation signals was used to reconstruct the full methylation maps of
the Neanderthal and the Denisovan.
In September 2019, researchers published a novel method to infer
morphological traits from DNA methylation data. The authors were able to
show that linking down-regulated genes to phenotypes of monogenic
diseases, where one or two copies of a gene are perturbed, allows for
~85% accuracy in reconstructing anatomical traits directly from DNA
methylation maps.
Methylation Sensitive Single Nucleotide Primer Extension Assay
(msSNuPE), which uses internal primers annealing straight 5' of the
nucleotide to be detected.
Illumina Methylation Assay
measures locus-specific DNA methylation using array hybridization.
Bisulfite-treated DNA is hybridized to probes on "BeadChips."
Single-base base extension with labeled probes is used to determine
methylation status of target sites.
In 2016, the Infinium MethylationEPIC BeadChip was released, which
interrogates over 850,000 methylation sites across the human genome.
Using nanopore sequencing, researchers have directly identified DNA
and RNA base modifications at nucleotide resolution, including 5mC,
5hmC, 6mA, and BrdU in DNA, and m6A in RNA, with detection of other
natural or synthetic epigenetic modifications possible through training
basecalling algorithms.
Differentially methylated regions (DMRs)
Differentially methylated regions,
are genomic regions with different methylation statuses among multiple
samples (tissues, cells, individuals or others), are regarded as
possible functional regions involved in gene transcriptional regulation.
The identification of DMRs among multiple tissues (T-DMRs) provides a
comprehensive survey of epigenetic differences among human tissues.
For example, these methylated regions that are unique to a particular
tissue allow individuals to differentiate between tissue type, such as
semen and vaginal fluid. Current research conducted by Lee et al.,
showed DACT1 and USP49 positively identified semen by examining T-DMRs.
The use of T-DMRs has proven useful in the identification of various
body fluids found at crime scenes. Researchers in the forensic field are
currently seeking novel T-DMRs in genes to use as markers in forensic
DNA analysis. DMRs between cancer and normal samples (C-DMRs)
demonstrate the aberrant methylation in cancers. It is well known that DNA methylation is associated with cell differentiation and proliferation. Many DMRs have been found in the development stages (D-DMRs) and in the reprogrammed progress (R-DMRs).
In addition, there are intra-individual DMRs (Intra-DMRs) with
longitudinal changes in global DNA methylation along with the increase
of age in a given individual. There are also inter-individual DMRs (Inter-DMRs) with different methylation patterns among multiple individuals.
QDMR (Quantitative Differentially Methylated Regions) is a
quantitative approach to quantify methylation difference and identify
DMRs from genome-wide methylation profiles by adapting Shannon entropy.
The platform-free and species-free nature of QDMR makes it potentially
applicable to various methylation data. This approach provides an
effective tool for the high-throughput identification of the functional
regions involved in epigenetic regulation. QDMR can be used as an
effective tool for the quantification of methylation difference and
identification of DMRs across multiple samples.
Gene-set analysis (a.k.a. pathway analysis; usually performed
tools such as DAVID, GoSeq or GSEA) has been shown to be severely biased
when applied to high-throughput methylation data (e.g. MeDIP-seq,
MeDIP-ChIP, HELP-seq etc.), and a wide range of studies have thus
mistakenly reported hyper-methylation of genes related to development
and differentiation; it has been suggested that this can be corrected
using sample label permutations or using a statistical model to control
for differences in the numbers of CpG probes / CpG sites that target
each gene.
DNA methylation marks
DNA methylation marks
– genomic regions with specific methylation patterns in a specific
biological state such as tissue, cell type, individual – are regarded as
possible functional regions involved in gene transcriptional
regulation. Although various human cell types may have the same genome,
these cells have different methylomes. The systematic identification and
characterization of methylation marks across cell types are crucial to
understanding the complex regulatory network for cell fate
determination. Hongbo Liu et al. proposed an entropy-based framework
termed SMART to integrate the whole genome bisulfite sequencing
methylomes across 42 human tissues/cells and identified 757,887 genome
segments.
Nearly 75% of the segments showed uniform methylation across all cell
types. From the remaining 25% of the segments, they identified cell
type-specific hypo/hypermethylation marks that were specifically
hypo/hypermethylated in a minority of cell types using a statistical
approach and presented an atlas of the human methylation marks. Further
analysis revealed that the cell type-specific hypomethylation marks were
enriched through H3K27ac
and transcription factor binding sites in a cell type-specific manner.
In particular, they observed that the cell type-specific hypomethylation
marks are associated with the cell type-specific super-enhancers that
drive the expression of cell identity genes. This framework provides a
complementary, functional annotation of the human genome and helps to
elucidate the critical features and functions of cell type-specific
hypomethylation.
The entropy-based Specific Methylation Analysis and Report Tool,
termed "SMART", which focuses on integrating a large number of DNA
methylomes for the de novo identification of cell type-specific
methylation marks. The latest version of SMART is focused on three main
functions including de novo identification of differentially methylated
regions (DMRs) by genome segmentation, identification of DMRs from
predefined regions of interest, and identification of differentially
methylated CpG sites.
In identification and detection of body fluids
DNA
methylation allows for several tissues to be analyzed in one assay as
well as for small amounts of body fluid to be identified with the use of
extracted DNA. Usually, the two approaches of DNA methylation are
either methylated-sensitive restriction enzymes or treatment with sodium
bisulphite.
Methylated sensitive restriction enzymes work by cleaving specific CpG,
cytosine and guanine separated by only one phosphate group, recognition
sites when the CpG is methylated. In contrast, unmethylated cytosines
are transformed to uracil and in the process, methylated cytosines
remain methylated. In particular, methylation profiles can provide
insight on when or how body fluids were left at crime scenes, identify
the kind of body fluid, and approximate age, gender, and phenotypic
characteristics of perpetrators.
Research indicates various markers that can be used for DNA
methylation. Deciding which marker to use for an assay is one of the
first steps of the identification of body fluids. In general, markers
are selected by examining prior research conducted. Identification
markers that are chosen should give a positive result for one type of
cell. One portion of the chromosome that is an area of focus when
conducting DNA methylation are tissue-specific differentially methylated
regions, T-DMRs. The degree of methylation for the T-DMRs ranges
depending on the body fluid.
A research team developed a marker system that is two-fold. The first
marker is methylated only in the target fluid while the second is
methylated in the rest of the fluids.
For instance, if venous blood marker A is un-methylated and venous
blood marker B is methylated in a fluid, it indicates the presence of
only venous blood. In contrast, if venous blood marker A is methylated
and venous blood marker B is un-methylated in some fluid, then that
indicates venous blood is in a mixture of fluids. Some examples for DNA
methylation markers are Mens1(menstrual blood), Spei1(saliva), and
Sperm2(seminal fluid).
DNA methylation provides a relatively good means of sensitivity
when identifying and detecting body fluids. In one study, only ten
nanograms of a sample was necessary to ascertain successful results.
DNA methylation provides a good discernment of mixed samples since it
involves markers that give “on or off” signals. DNA methylation is not
impervious to external conditions. Even under degraded conditions using
the DNA methylation techniques, the markers are stable enough that there
are still noticeable differences between degraded samples and control
samples. Specifically, in one study, it was found that there were not
any noticeable changes in methylation patterns over an extensive period
of time.
Computational prediction
DNA
methylation can also be detected by computational models through
sophisticated algorithms and methods. Computational models can
facilitate the global profiling of DNA methylation across chromosomes,
and often such models are faster and cheaper to perform than biological
assays. Such up-to-date computational models include Bhasin, et al., Bock, et al., and Zheng, et al. Together with biological assay, these methods greatly facilitate the DNA methylation analysis.









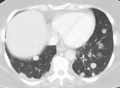
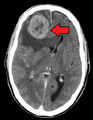







Mark.png)










.png)



