Immunosenescence involves both the host's capacity to respond to
infections and the development of long-term immune memory, especially by
vaccination. This age-associated immune deficiency
is ubiquitous and found in both long- and short-living species as a
function of their age relative to life expectancy rather than
chronological time. It is considered a major contributory factor to the increased frequency of morbidity and mortality among the elderly.
Immunosenescence is not a random deteriorative phenomenon, rather
it appears to inversely repeat an evolutionary pattern and most of the
parameters affected by immunosenescence appear to be under genetic
control.
Immunosenescence can also be sometimes envisaged as the result of the
continuous challenge of the unavoidable exposure to a variety of antigens such as viruses and bacteria.
Overview of the age-associated decline in immune function
Immunosenescence
is a multifactorial condition leading to many pathologically
significant health problems in the aged population. Some of the
age-dependent biological changes that contribute to the onset of
immunosenescence are listed below:
Hematopoietic stem cells (HSC), which provide the regulated lifelong supply of leukocyte progenitors that are in turn able to differentiate into a diversity of specialised immune cells (including lymphocytes, antigen-presentingdendritic cells and phagocytes) diminish in their self-renewal capacity. This is due to the accumulation of oxidative damage to DNA by aging and cellular metabolic activity and the shortening of telomeric terminals of chromosomes.
There is a notable decline in the total number of phagocytes in aged hosts, coupled with an intrinsic reduction of their bactericidal activity.
The cytotoxicity of Natural Killer (NK) cells and the antigen-presenting function of dendritic cells is known to diminish with old age. The age-associated impairment of dendritic Antigen Presenting Cells (APCs) has profound implications as this translates into a deficiency in cell-mediated immunity and thus, the inability for effector T-lymphocytes to modulate an adaptive immune response (see below).
As age advances, there is decline in both the production of new naive lymphocytes
and the functional competence of memory cell populations. This has been
implicated in the increasing frequency and severity of diseases such as
cancer, chronic inflammatory disorders, breakthrough infections and autoimmunity. A problem of infections in the elderly is that they frequently present
with non-specific signs and symptoms, and clues of focal infection are
often absent or obscured by underlying chronic conditions. Ultimately, this provides problems in diagnosis and subsequently, treatment.
In addition to changes in immune responses, the beneficial effects of inflammation
devoted to the neutralisation of dangerous and harmful agents early in
life and in adulthood become detrimental late in life in a period
largely not foreseen by evolution, according to the antagonistic pleiotropy theory of aging. It should be further noted that changes in the lymphoid compartment is not solely responsible for the malfunctioning of the immune system in the elderly. Although myeloid cell production does not seem to decline with age, macrophages become dysregulated as a consequence of environmental changes.
T-cell functional dysregulation as a biomarker for immunosenescence
The functional capacity of T-cells
is most influenced by the effects of aging. In fact, age-related
alterations are evident in all stages of T-cell development, making them
a significant factor in the development of immunosenescence. After birth, the decline of T-cell function begins with the progressive involution of the thymus,
which is the organ essential for T-cell maturation following the
migration of precursor cells from the bone marrow. This age-associated
decrease of thymic epithelial volume results in a reduction/exhaustion on the number of thymocytes (i.e. pre-mature T-cells), thus reducing output of peripheral naïve T-cells.
Once matured and circulating throughout the peripheral system, T-cells
still undergo deleterious age-dependent changes. Together with the
age-related thymic involution, and the consequent age-related decrease
of thymic output of new T cells, this situation leaves the body
practically devoid of virgin T cells, which makes the body more prone to
a variety of infectious and non-infectious diseases.
By age 40, and estimated 50% to 85% of adults have contracted human cytomegalivirus (HCMV), which is believed to be a major cause of immunosenescence, although this is controversial.[21] Despite the fact that an average of 10% (and up to 50%) of the CD4 and CD8memory T cells of HCMV-infected persons may be CMV-specific, these persons do not have a higher fatality rate resulting from other infections.
T-cell components associated with immunosenescence include:
A T cell is a type of lymphocyte, which develops in the thymus gland (hence the name) and plays a central role in the immune response. T cells can be distinguished from other lymphocytes by the presence of a T-cell receptor on the cell surface. These immune cells originate as precursor cells, derived from bone marrow,
and develop into several distinct types of T cells once they have
migrated to the thymus gland. T cell differentiation continues even
after they have left the thymus.
Groups of specific, differentiated T cells have an important role
in controlling and shaping the immune response by providing a variety
of immune-related functions. One of these functions is immune-mediated
cell death, and it is carried out by T cells in several ways: CD8+ T cells, also known as "killer cells", are cytotoxic
- this means that they are able to directly kill virus-infected cells
as well as cancer cells. CD8+ T cells are also able to utilize small
signalling proteins, known as cytokines, to recruit other cells when mounting an immune response. A different population of T cells, the CD4+
T cells, function as "helper cells". Unlike CD8+ killer T cells, these
CD4+ helper T cells function by indirectly killing cells identified as
foreign: they determine if and how other parts of the immune system
respond to a specific, perceived threat. Helper T cells also use
cytokine signalling to influence regulatory B cells directly, and other cell populations indirectly. Regulatory T cells are yet another distinct population of these cells that provide the critical mechanism of tolerance,
whereby immune cells are able to distinguish invading cells from "self"
- thus preventing immune cells from inappropriately mounting a response
against oneself (which would by definition be an "autoimmune"
response). For this reason these regulatory T cells have also been
called "suppressor" T cells. These same self-tolerant cells are
co-opted by cancer cells to prevent the recognition of, and an immune
response against, tumour cells.
Development
Origin, early development and migration to the thymus
All T cells originate from c-kit+Sca1+haematopoietic stem cells
(HSC) which reside in the bone marrow. In some cases, the origin might
be the fetal liver during embryonic development. The HSC then
differentiate into multipotent progenitors (MPP) which retain the
potential to become both myeloid and lymphoid cells. The process of
differentiation then proceeds to a common lymphoid progenitor (CLP),
which can only differentiate into T, B or NK cells.
These CLP cells then migrate via the blood to the thymus, where they
engraft. The earliest cells which arrived in the thymus are termed
double-negative, as they express neither the CD4 nor CD8 co-receptor. The newly arrived CLP cells are CD4-CD8-CD44+CD25-ckit+ cells, and are termed early thymic progenitors (ETP) cells. These cells will then undergo a round of division and downregulate c-kit and are termed DN1 cells.
TCR-Beta selection
At the DN2 stage (CD44+CD25+),
cells upregulate the recombination genes RAG1 and RAG2 and re-arrange
the TCRβ locus, combining V-D-J and constant region genes in an attempt
to create a functional TCRβ chain. As the developing thymocyte
progresses through to the DN3 stage (CD44-CD25+),
the T cell expresses an invariant α-chain called pre-Tα alongside the
TCRβ gene. If the rearranged β-chain successfully pairs with the
invariant α-chain, signals are produced which cease rearrangement of the
β-chain (and silences the alternate allele).
Although these signals require this pre-TCR at the cell surface, they
are independent of ligand binding to the pre-TCR. If the pre-TCR forms,
then the cell downregulates CD25 and is termed a DN4 cell (CD25-CD44-). These cells then undergo a round of proliferation and begin to re-arrange the TCRα locus.
Positive selection
Double-positive thymocytes (CD4+/CD8+) migrate deep into the thymic cortex, where they are presented with self-antigens. These self-antigens are expressed by thymic cortical epithelial cells on MHC
molecules on the surface of cortical epithelial cells. Only those
thymocytes that interact with MHC-I or MHC-II will receive a vital
"survival signal". All that cannot (if they do not interact strongly
enough) will die by "death by neglect" (no survival signal). This
process ensures that the selected T cells will have an MHC affinity that
can serve useful functions in the body (i.e., the cells must be able to
interact with MHC and peptide complexes to effect immune responses).
The vast majority of developing thymocytes will die during this process.
The process of positive selection takes a number of days.
A thymocyte's fate is determined during positive selection. Double-positive cells (CD4+/CD8+) that interact well with MHC class II molecules will eventually become CD4+ cells, whereas thymocytes that interact well with MHC class I molecules mature into CD8+ cells. A T cell becomes a CD4+
cell by down-regulating expression of its CD8 cell surface receptors.
If the cell does not lose its signal, it will continue downregulating
CD8 and become a CD4+, single positive cell.
This process does not remove thymocytes that may cause autoimmunity.
The potentially autoimmune cells are removed by the process of negative
selection, which occurs in the thymic medulla (discussed below).
Negative selection
Negative
selection removes thymocytes that are capable of strongly binding with
"self" MHC peptides. Thymocytes that survive positive selection migrate
towards the boundary of the cortex and medulla in the thymus. While in
the medulla, they are again presented with a self-antigen presented on
the MHC complex of medullary thymic epithelial cells (mTECs). mTECs must be AIRE+
to properly express self-antigens from all tissues of the body on their
MHC class I peptides. Some mTECs are phagocytosed by thymic dendritic
cells; this allows for presentation of self-antigens on MHC class II
molecules (positively selected CD4+ cells must interact with MHC class II molecules, thus APCs, which possess MHC class II, must be present for CD4+ T-cell negative selection). Thymocytes that interact too strongly with the self-antigen receive an apoptotic signal that leads to cell death. However, some of these cells are selected to become Treg cells. The remaining cells exit the thymus as mature naïve T cells (also known as recent thymic emigrants). This process is an important component of central tolerance and serves to prevent the formation of self-reactive T cells that are capable of inducing autoimmune diseases in the host.
β-selection is the first checkpoint, where the T cells that are
able to form a functional pre-TCR with an invariant alpha chain and a
functional beta chain are allowed to continue development in the thymus.
Next, positive selection checks that T cells have successfully
rearranged their TCRα locus and are capable of recognizing peptide-MHC
complexes with appropriate affinity. Negative selection in the medulla
then obliterates T cells that bind too strongly to self-antigens
expressed on MHC molecules. These selection processes allow for
tolerance of self by the immune system. Typical T cells that leave the
thymus (via the corticomedullary junction) are self-restricted,
self-tolerant, and single positive.
Thymic output
About
98% of thymocytes die during the development processes in the thymus by
failing either positive selection or negative selection, whereas the
other 2% survive and leave the thymus to become mature immunocompetent T
cells.
The thymus contributes fewer cells as a person ages. As the thymus
shrinks by about 3% a year throughout middle age, a corresponding fall in the thymic production of naïve T cells occurs, leaving peripheral T cell expansion and regeneration to play a greater role in protecting older people.
Types of T cell
T
cells are grouped into a series of subsets based on their function. CD4
and CD8 T cells are selected in the thymus, but undergo further
differentiation in the periphery to specialized cells which have
different functions. T cell subsets were initially defined by function,
but also have associated gene or protein expression patterns.
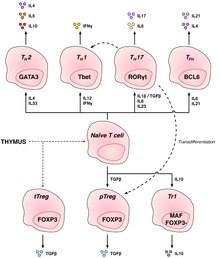
Depiction of the various key subsets of CD4-positive T cells with corresponding associated cytokines and transcription factors.
Conventional Adaptive T cells
Helper CD4+ T cells
T helper cells (TH cells) assist other lymphocytes, including maturation of B cells into plasma cells and memory B cells, and activation of cytotoxic T cells and macrophages. These cells are also known as CD4+ T cells as they express the CD4 on their surfaces. Helper T cells become activated when they are presented with peptideantigens by MHC class II molecules, which are expressed on the surface of antigen-presenting cells (APCs). Once activated, they divide rapidly and secrete cytokines
that regulate or assist the immune response. These cells can
differentiate into one of several subtypes, which have different roles.
Cytokines direct T cells into particular subtypes.
Superresolution image of a group of cytotoxic T cells surrounding a cancer cell
Cytotoxic T cells (TC cells, CTLs, T-killer cells, killer T cells) destroy virus-infected cells and tumor cells, and are also implicated in transplant rejection. These cells are defined by the expression of CD8+ on the cell surface. These cells recognize their targets by binding to short peptides (8-11AA) associated with MHC class I
molecules, present on the surface of all nucleated cells. CD8+ T cells
also produce the key cytokines IL-2 and IFNγ, which influence the
effector functions of other cells, in particular macrophages and NK
cells.
Memory T cells
Antigen-naïve T cells expand and differentiate into memory and effector T cells,
after they encounter their cognate antigen within the context of an MHC
molecule on the surface of a professional antigen presenting cell (e.g.
a dendritic cell). Appropriate co-stimulation must be present at the
time of antigen encounter for this process to occur. Historically,
memory T cells were thought to belong to either the effector or central
memory subtypes, each with their own distinguishing set of cell surface
markers (see below).
Subsequently, numerous new populations of memory T cells were
discovered including tissue-resident memory T (Trm) cells, stem memory
TSCM cells, and virtual memory T cells. The single unifying theme for
all memory T cell
subtypes is that they are long-lived and can quickly expand to large
numbers of effector T cells upon re-exposure to their cognate antigen.
By this mechanism they provide the immune system with "memory" against
previously encountered pathogens. Memory T cells may be either CD4+ or CD8+ and usually express CD45RO.
Memory T cell subtypes:
Central memory T cells (TCM cells) express CD45RO, C-C chemokine receptor type 7 (CCR7), and L-selectin (CD62L). Central memory T cells also have intermediate to high expression of CD44. This memory subpopulation is commonly found in the lymph nodes and in the peripheral circulation. (Note- CD44 expression is usually used to distinguish murine naive from memory T cells).
Effector memory T cells (TEM cells and TEMRA cells) express CD45RO but lack expression of CCR7 and L-selectin. They also have intermediate to high expression of CD44. These memory T cells lack lymph node-homing receptors and are thus found in the peripheral circulation and tissues. TEMRA
stands for terminally differentiated effector memory cells
re-expressing CD45RA, which is a marker usually found on naive T cells.
Tissue resident memory T cells (TRM) occupy tissues (skin, lung, etc..) without recirculating. One cell surface marker that has been associated with TRM is the intern αeβ7, also known as CD103.
Virtual memory T cells differ from the other memory subsets in that
they do not originate following a strong clonal expansion event. Thus,
although this population as a whole is abundant within the peripheral
circulation, individual virtual memory T cell clones reside at
relatively low frequencies. One theory is that homeostatic proliferation
gives rise to this T cell population. Although CD8 virtual memory T
cells were the first to be described, it is now known that CD4 virtual memory cells also exist.
Regulatory CD4+ T cells
Regulatory T cells are crucial for the maintenance of immunological tolerance. Their major role is to shut down T cell-mediated immunity toward the end of an immune reaction and to suppress autoreactive T cells that escaped the process of negative selection in the thymus.
Two major classes of CD4+ Treg cells have been described — FOXP3+ Treg cells and FOXP3− Treg cells.
Regulatory T cells can develop either during normal development
in the thymus, and are then known as thymic Treg cells, or can be
induced peripherally and are called peripherally derived Treg cells.
These two subsets were previously called "naturally occurring", and
"adaptive" or "induced", respectively. Both subsets require the expression of the transcription factorFOXP3 which can be used to identify the cells. Mutations of the FOXP3 gene can prevent regulatory T cell development, causing the fatal autoimmune diseaseIPEX.
Several other types of T cell have suppressive activity, but do
not express FOXP3. These include Tr1 cells and Th3 cells, which are
thought to originate during an immune response and act by producing
suppressive molecules. Tr1 cells are associated with IL-10, and Th3 cells are associated with TGF-beta. Recently, Treg17 cells have been added to this list.
Innate-like T cells
Natural killer T cell
Natural killer T cells (NKT cells – not to be confused with natural killer cells of the innate immune system) bridge the adaptive immune system with the innate immune system. Unlike conventional T cells that recognize peptide antigens presented by major histocompatibility complex (MHC) molecules, NKT cells recognize glycolipid antigen presented by CD1d. Once activated, these cells can perform functions ascribed to both Th and Tc
cells (i.e., cytokine production and release of cytolytic/cell killing
molecules). They are also able to recognize and eliminate some tumor
cells and cells infected with herpes viruses.
Mucosal associated invariant
MAIT cells display innate, effector-like qualities. In humans, MAIT cells are found in the blood, liver, lungs, and mucosa, defending against microbial activity and infection. The MHC class I-like protein, MR1, is responsible for presenting bacterially-produced vitamin B metabolites to MAIT cells. After the presentation of foreign antigen by MR1, MAIT cells secretes pro-inflammatory cytokines and are capable of lysing bacterially-infected cells. MAIT cells can also be activated through MR1-independent signaling. In addition to possessing innate-like functions, this T cell subset supports the adaptive immune response and has a memory-like phenotype. Furthermore, MAIT cells are thought to play a role in autoimmune diseases, such as multiple sclerosis, arthritis and inflammatory bowel disease, although definitive evidence is yet to be published.
Gamma delta T cells
Gamma delta T cells
(γδ T cells) represent a small subset of T cells which possess a γδ TCR
rather than the αβ TCR on the cell surface. The majority of T cells
express αβ TCR chains. This group of T cells is much less common in
humans and mice (about 2% of total T cells) and are found mostly in the
gut mucosa, within a population of intraepithelial lymphocytes.
In rabbits, sheep, and chickens, the number of γδ T cells can be as
high as 60% of total T cells. The antigenic molecules that activate γδ T
cells are still mostly unknown. However, γδ T cells are not
MHC-restricted and seem to be able to recognize whole proteins rather
than requiring peptides to be presented by MHC molecules on APCs. Some murine
γδ T cells recognize MHC class IB molecules. Human γδ T cells which
use the Vγ9 and Vδ2 gene fragments constitute the major γδ T cell
population in peripheral blood, and are unique in that they specifically
and rapidly respond to a set of nonpeptidic phosphorylated isoprenoid precursors, collectively named phosphoantigens,
which are produced by virtually all living cells. The most common
phosphoantigens from animal and human cells (including cancer cells) are
isopentenyl pyrophosphate (IPP) and its isomer dimethylallyl pyrophosphate (DMPP). Many microbes produce the highly active compound hydroxy-DMAPP (HMB-PP)
and corresponding mononucleotide conjugates, in addition to IPP and
DMAPP. Plant cells produce both types of phosphoantigens. Drugs
activating human Vγ9/Vδ2 T cells comprise synthetic phosphoantigens and aminobisphosphonates, which upregulate endogenous IPP/DMAPP.
Activation
The
T lymphocyte activation pathway: T cells contribute to immune defenses
in two major ways; some direct and regulate immune responses; others
directly attack infected or cancerous cells.
Activation of CD4+ T cells occurs through the simultaneous engagement of the T-cell receptor and a co-stimulatory molecule (like CD28, or ICOS) on the T cell by the major histocompatibility complex (MHCII) peptide
and co-stimulatory molecules on the APC. Both are required for
production of an effective immune response; in the absence of co-stimulation, T cell receptor signalling alone results in anergy. The signalling pathways downstream from co-stimulatory molecules usually engages the PI3K pathway generating PIP3 at the plasma membrane and recruiting PH domain containing signaling molecules like PDK1 that are essential for the activation of PKCθ, and eventual IL-2 production. Optimal CD8+ T cell response relies on CD4+ signalling. CD4+ cells are useful in the initial antigenic activation of naïve CD8 T cells, and sustaining memory CD8+ T cells in the aftermath of an acute infection. Therefore, activation of CD4+ T cells can be beneficial to the action of CD8+ T cells.
The first signal is provided by binding of the T cell receptor to
its cognate peptide presented on MHCII on an APC. MHCII is restricted
to so-called professional antigen-presenting cells, like dendritic cells, B cells, and macrophages, to name a few. The peptides presented to CD8+ T cells by MHC class I molecules are 8–13 amino acids in length; the peptides presented to CD4+ cells by MHC class II molecules are longer, usually 12–25 amino acids in length, as the ends of the binding cleft of the MHC class II molecule are open.
The second signal comes from co-stimulation, in which surface
receptors on the APC are induced by a relatively small number of
stimuli, usually products of pathogens, but sometimes breakdown products
of cells, such as necrotic-bodies or heat shock proteins.
The only co-stimulatory receptor expressed constitutively by naïve T
cells is CD28, so co-stimulation for these cells comes from the CD80 and CD86 proteins, which together constitute the B7 protein, (B7.1 and B7.2, respectively) on the APC. Other receptors are expressed upon activation of the T cell, such as OX40
and ICOS, but these largely depend upon CD28 for their expression. The
second signal licenses the T cell to respond to an antigen. Without
it, the T cell becomes anergic,
and it becomes more difficult for it to activate in future. This
mechanism prevents inappropriate responses to self, as self-peptides
will not usually be presented with suitable co-stimulation. Once a T
cell has been appropriately activated (i.e. has received signal one and
signal two) it alters its cell surface expression of a variety of
proteins. Markers of T cell activation include CD69, CD71 and CD25
(also a marker for Treg cells), and HLA-DR (a marker of human T cell
activation). CTLA-4 expression is also up-regulated on activated T
cells, which in turn outcompetes CD28 for binding to the B7 proteins.
This is a checkpoint mechanism to prevent over activation of the T cell.
Activated T cells also change their cell surface glycosylation profile.
The T cell receptor
exists as a complex of several proteins. The actual T cell receptor is
composed of two separate peptide chains, which are produced from the
independent T cell receptor alpha and beta (TCRα and TCRβ) genes. The other proteins in the complex are the CD3 proteins: CD3εγ and CD3εδ heterodimers and, most important, a CD3ζ homodimer, which has a total of six ITAM motifs. The ITAM motifs on the CD3ζ can be phosphorylated by Lck and in turn recruit ZAP-70. Lck and/or ZAP-70 can also phosphorylate the tyrosines on many other molecules, not least CD28, LAT and SLP-76, which allows the aggregation of signalling complexes around these proteins.
Phosphorylated LAT recruits SLP-76 to the membrane, where it can then bring in PLC-γ, VAV1, Itk and potentially PI3K. PLC-γ cleaves PI(4,5)P2 on the inner leaflet of the membrane to create the active intermediaries diacylglycerol (DAG), inositol-1,4,5-trisphosphate (IP3);
PI3K also acts on PIP2, phosphorylating it to produce
phosphatidlyinositol-3,4,5-trisphosphate (PIP3). DAG binds and
activates some PKCs. Most important in T cells is PKCθ, critical for
activating the transcription factors NF-κB and AP-1. IP3 is released from the membrane by PLC-γ and diffuses rapidly to activate calcium channel receptors on the ER, which induces the release of calcium
into the cytosol. Low calcium in the endoplasmic reticulum causes
STIM1 clustering on the ER membrane and leads to activation of cell
membrane CRAC channels that allows additional calcium to flow into the
cytosol from the extracellular space. This aggregated cytosolic calcium
binds calmodulin, which can then activate calcineurin. Calcineurin, in turn, activates NFAT, which then translocates to the nucleus. NFAT is a transcription factor
that activates the transcription of a pleiotropic set of genes, most
notable, IL-2, a cytokine that promotes long-term proliferation of
activated T cells.
PLCγ can also initiate the NF-κB pathway.
DAG activates PKCθ, which then phosphorylates CARMA1, causing it to
unfold and function as a scaffold. The cytosolic domains bind an
adapter BCL10 via CARD (Caspase activation and recruitment domains) domains; that then binds TRAF6, which is ubiquitinated at K63.
This form of ubiquitination does not lead to degradation of target
proteins. Rather, it serves to recruit NEMO, IKKα and -β, and TAB1-2/
TAK1.
TAK 1 phosphorylates IKK-β, which then phosphorylates IκB allowing for
K48 ubiquitination: leads to proteasomal degradation. Rel A and p50
can then enter the nucleus and bind the NF-κB response element. This
coupled with NFAT signaling allows for complete activation of the IL-2
gene.
While in most cases activation is dependent on TCR recognition of
antigen, alternative pathways for activation have been described. For
example, cytotoxic T cells have been shown to become activated when
targeted by other CD8 T cells leading to tolerization of the latter.
A
unique feature of T cells is their ability to discriminate between
healthy and abnormal (e.g. infected or cancerous) cells in the body.
Healthy cells typically express a large number of self derived pMHC on
their cell surface and although the T cell antigen receptor can interact
with at least a subset of these self pMHC, the T cell generally ignores
these healthy cells. However, when these very same cells contain even
minute quantities of pathogen derived pMHC, T cells are able to become
activated and initiate immune responses. The ability of T cells to
ignore healthy cells but respond when these same cells contain pathogen
(or cancer) derived pMHC is known as antigen discrimination. The
molecular mechanisms that underlie this process are controversial.
T
cell exhaustion is a state of dysfunctional T cells. It is
characterized by progressive loss of function, changes in
transcriptional profiles and sustained expression of inhibitory
receptors. At first cells lose their ability to produce IL-2 and TNFα
followed by the loss of high proliferative capacity and cytotoxic
potential, eventually leading to their deletion. Exhausted T cells
typically indicate higher levels of CD43, CD69 and inhibitory receptors combined with lower expression of CD62L and CD127. Exhaustion can develop during chronic infections, sepsis and cancer. Exhausted T cells preserve their functional exhaustion even after repeated antigen exposure.
During chronic infection and sepsis
T cell exhaustion can be triggered by several factors like persistent antigen exposure and lack of CD4 T cell help.
Antigen exposure also has effect on the course of exhaustion because
longer exposure time and higher viral load increases the severity of T
cell exhaustion. At least 2–4 weeks exposure is needed to establish
exhaustion. Another factor able to induce exhaustion are inhibitory receptors including programmed cell death protein 1 (PD1), CTLA-4, T cell membrane protein-3 (TIM3), and lymphocyte activation gene 3 protein (LAG3). Soluble molecules such as cytokines IL-10 or TGF-β are also able to trigger exhaustion. Last known factors that can play a role in T cell exhaustion are regulatory cells. Treg cells can be a source of IL-10 and TGF-β and therefore they can play a role in T cell exhaustion. Furthermore, T cell exhaustion is reverted after depletion of Treg cells and blockade of PD1.
T cell exhaustion can also occur during sepsis as a result of cytokine
storm. Later after the initial septic encounter anti-inflammatory
cytokines and pro-apoptotic proteins take over to protect the body from
damage. Sepsis also carries high antigen load and inflammation. In this
stage of sepsis T cell exhaustion increases. Currently there are studies aiming to utilize inhibitory receptor blockades in treatment of sepsis.
During transplantation
While
during infection T cell exhaustion can develop following persistent
antigen exposure after graft transplant similar situation arises with
alloantigen presence. It was shown that T cell response diminishes over time after kidney transplant.
These data suggest T cell exhaustion plays an important role in
tolerance of a graft mainly by depletion of alloreactive CD8 T cells.
Several studies showed positive effect of chronic infection on graft
acceptance and its long-term survival mediated partly by T cell
exhaustion. It was also shown that recipient T cell exhaustion provides sufficient conditions for NK cell transfer.
While there are data showing that induction of T cell exhaustion can be
beneficial for transplantation it also carries disadvantages among
which can be counted increased number of infections and the risk of
tumor development.
During cancer
During
cancer T cell exhaustion plays a role in tumor protection. According to
research some cancer-associated cells as well as tumor cells themselves
can actively induce T cell exhaustion at the site of tumor. T cell exhaustion can also play a role in cancer relapses as was shown on leukemia.
Some study even suggested that it is possible to predict relapse of
leukemia based on expression of inhibitory receptors PD-1 and TIM-3 by T
cells.
In recent years there is a lot of experiments and clinical trials with
immune checkpoint blockers in cancer therapy. Some of them were approved
as valid therapies and are now used in clinics.
Inhibitory receptors targeted by those medical procedures are vital in T
cell exhaustion and blocking them can reverse these changes.
In medicine, a side effect
is an effect, whether therapeutic or adverse, that is secondary to the
one intended; although the term is predominantly employed to describe adverse effects, it can also apply to beneficial, but unintended, consequences of the use of a drug. Developing drugs
is a complicated process, because no two people are exactly the same,
so even drugs that have virtually no side effects, might be difficult
for some people. Also, it is difficult to make a drug that targets one
part of the body but that doesn't affect other parts, the fact that increases the risk of side effects in the untargeted parts.
Occasionally, drugs are prescribed or procedures performed
specifically for their side effects; in that case, said side effect
ceases to be a side effect, and is now an intended effect. For
instance, X-rays
were historically (and are currently) used as an imaging technique; the
discovery of their oncolytic capability led to their employ in radiotherapy (ablation of malignanttumours).
Frequency of side effects
The probability or chance of experiencing side effects are characterised as :
Buprenorphine has been shown experimentally (1982–1995) to be effective against severe, refractory depression.
Bupropion (Wellbutrin), an anti-depressant,
is also used as a smoking cessation aid; this indication was later
approved, and the name of the smoking cessation product is Zyban. In
Ontario, Canada, smoking cessation drugs are not covered by provincial
drug plans; elsewhere, Zyban is priced higher than Wellbutrin, despite
being the same drug. Therefore, some physicians prescribe Wellbutrin for
both indications.
The SSRI medication sertraline is approved as an antidepressant but delays conjugal climax in men, and thus may be supplied to those in which climax is premature.
Terazosin, an α1-adrenergic antagonist
approved to treat benign prostatic hyperplasia (enlarged prostate) and
hypertension, is (one of several drugs) used off-label to treat drug
induced diaphoresis and hyperhidrosis (excessive sweating).
Examples of undesirable/unwanted side effects
Echinacea
– more than 20 different types of reactions have been reported,
including asthma attacks, loss of pregnancy, hives, swelling, aching
muscles and gastrointestinal upsets.
Feverfew
– pregnant women should avoid using this herb, as it can trigger
uterine contractions which could lead to premature labour or
miscarriage.
Antimalarial medications or simply antimalarials are a type of antiparasitic chemical agent, often naturally derived, that can be used to treat or to prevent malaria, in the latter case, most often aiming at two susceptible target groups, young children and pregnant women. As of 2018, modern treatments, including for severe malaria, continued to depend on therapies deriving historically from quinine and artesunate, both parenteral (injectable) drugs, expanding from there into the many classes of available modern drugs.
Incidence and distribution of the disease ("malaria burden") is
expected to remain high, globally, for many years to come; moreover,
known antimalarial drugs have repeatedly been observed to elicit
resistance in the malaria parasite—including for combination therapies
featuring artemisinin, a drug of last resort, where resistance has now been observed in Southeast Asia.
As such, the needs for new antimalarial agents and new strategies of
treatment (e.g., new combination therapies) remain important priorities
in tropical medicine. As well, despite very positive outcomes from many modern treatments, serious side effects can impact some individuals taking standard doses (e.g., retinopathy with chloroquine, acute haemolytic anaemia with tafenoquine).
Specifically, antimalarial drugs may be used to treat malaria in
three categories of individuals, (i) those with suspected or confirmed
infection, (ii) those visiting a malaria-endemic regions who have no
immunity, to prevent infection via malaria prophylaxis,
and (iii) or in broader groups of individuals, in routine but
intermittent preventative treatment in regions where malaria is endemic
via intermittent preventive therapy. As of this date, practice in treating cases of malaria is most often based on the concept of combination therapy (e.g., using agents such as artemether and lumefantrine against chloroquine-resistant Plasmodium falciparum infection),
since this offers advantages including reduced risk of treatment
failure, reduced risk of developed resistance, as well as the
possibility of reduced side-effects.
Prompt parasitological confirmation by microscopy, or alternatively by
rapid diagnostic tests, is recommended in all patients suspected of
malaria before treatment is started. Treatment solely on the basis of clinical suspicion is considered when a parasitological diagnosis is not possible.
Medications
It
is practical to consider antimalarials by chemical structure since this
is associated with important properties of each drug, such as mechanism
of action.
Quinine and related agents
Quinine has a long history stretching from Peru, and the discovery of the cinchona tree, and the potential uses of its bark, to the current day and a collection of derivatives that are still frequently used in the prevention and treatment of malaria. Quinine is an alkaloid that acts as a blood schizonticidal and weak gametocide against Plasmodium vivax and Plasmodium malariae. As an alkaloid, it is accumulated in the food vacuoles of Plasmodium species, especially Plasmodium falciparum. It acts by inhibiting the hemozoinbiocrystallization, thus facilitating an aggregation of cytotoxic heme. Quinine is less effective and more toxic as a blood schizonticidal agent than chloroquine; however, it is still very effective and widely used in the treatment of acute cases of severe P. falciparum. It is especially useful in areas where there is known to be a high level of resistance to chloroquine, mefloquine, and sulfa drug combinations with pyrimethamine. Quinine is also used in post-exposure treatment of individuals returning from an area where malaria is endemic.
The treatment regimen of quinine is complex and is determined
largely by the parasite's level of resistance and the reason for drug
therapy (i.e. acute treatment or prophylaxis). The World Health Organization
recommendation for quinine is 20 mg/kg first times and 10 mg/kg every
eight hours for five days where parasites are sensitive to quinine,
combined with doxycycline, tetracycline or clindamycin. Doses can be given by oral, intravenous or intramuscular
routes. The recommended method depends on the urgency of treatment and
the available resources (i.e. sterilised needles for IV or IM
injections).
Use of quinine is characterised by a frequently experienced syndrome called cinchonism. Tinnitus (a hearing impairment), rashes, vertigo,
nausea, vomiting and abdominal pain are the most common symptoms.
Neurological effects are experienced in some cases due to the drug's neurotoxic properties. These actions are mediated through the interactions of quinine causing a decrease in the excitability of the motor neuronend plates. This often results in functional impairment of the eighth cranial nerve, resulting in confusion, delirium and coma. Quinine can cause hypoglycaemia through its action of stimulating insulin
secretion; this occurs in therapeutic doses and therefore it is advised
that glucose levels are monitored in all patients every 4–6 hours. This
effect can be exaggerated in pregnancy and therefore additional care in
administering and monitoring the dosage is essential. Repeated or
over-dosage can result in renal failure and death through depression of the respiratory system.
Quinimax and quinidine
are the two most commonly used alkaloids related to quinine in the
treatment or prevention of malaria. Quinimax is a combination of four
alkaloids (quinine, quinidine, cinchoine and cinchonidine). This
combination has been shown in several studies to be more effective than
quinine, supposedly due to a synergistic action between the four
cinchona derivatives. Quinidine is a direct derivative of quinine. It is
a distereoisomer,
thus having similar anti-malarial properties to the parent compound.
Quinidine is recommended only for the treatment of severe cases of
malaria.
Warburg's tincture was a febrifuge developed by Carl Warburg
in 1834, which included quinine as a key ingredient. In the
19th-century it was a well-known anti-malarial drug. Although originally
sold as a secret medicine, Warburg's tincture was highly regarded by
many eminent medical professionals who considered it as being superior
to quinine (e.g. Surgeon-General W. C. Maclean, Professor of Military
Medicine at British Army Medical School, Netley). Warburg's tincture
appeared in Martindale: The complete drug reference from 1883 until about 1920. The formula was published in The Lancet 1875.
Chloroquine
Chloroquine
was, until recently, the most widely used anti-malarial. It was the
original prototype from which most methods of treatment are derived. It
is also the least expensive, best tested and safest of all available
drugs. The emergence of drug-resistant parasitic strains is rapidly
decreasing its effectiveness; however, it is still the first-line drug
of choice in most sub-Saharan African
countries. It is now suggested that it is used in combination with
other antimalarial drugs to extend its effective usage. Popular drugs
based on chloroquine phosphate (also called nivaquine) are Chloroquine
FNA, Resochin and Dawaquin.
Chloroquine is a 4-aminoquinolone
compound with a complicated and still unclear mechanism of action. It
is believed to reach high concentrations in the vacuoles of the
parasite, which, due to its alkaline nature, raises the internal pH. It controls the conversion of toxic heme to hemozoin by inhibiting the biocrystallization of hemozoin,
thus poisoning the parasite through excess levels of toxicity. Other
potential mechanisms through which it may act include interfering with
the biosynthesis of parasitic nucleic acids and the formation of a chloroquine-haem or chloroquine-DNA
complex. The most significant level of activity found is against all
forms of the schizonts (with the obvious exception of
chloroquine-resistant P. falciparum and P. vivax strains) and the gametocytes of P. vivax, P. malariae, P. ovale as well as the immature gametocytes of P. falciparum. Chloroquine also has a significant anti-pyretic and anti-inflammatory effect when used to treat P. vivax
infections, and thus it may still remain useful even when resistance is
more widespread. According to a report on the Science and Development
Network website's sub-Saharan Africa section, there is very little drug
resistance among children infected with malaria on the island of
Madagascar, but what drug resistance there is exists against
chloroquinine.
Children and adults should receive 25 mg of chloroquine per kg given over three days. A pharmacokinetically
superior regime, recommended by the WHO, involves giving an initial
dose of 10 mg/kg followed 6–8 hours later by 5 mg/kg, then 5 mg/kg on
the following two days. For chemoprophylaxis: 5 mg/kg/week (single dose) or 10 mg/kg/week divided into six daily doses is advised. Chloroquine is only recommended as a prophylactic drug in regions only affected by P. vivax and sensitive P. falciparum strains. Chloroquine has been used in the treatment of malaria for many years and no abortifacient or teratogenic effects have been reported during this time; therefore, it is considered very safe to use during pregnancy. However, itching can occur at intolerable level and Chloroquinine can be a provocation factor of psoriasis.
Amodiaquine
Amodiaquine
is a 4-aminoquinolone anti-malarial drug similar in structure and
mechanism of action to chloroquine. Amodiaquine has tended to be
administered in areas of chloroquine resistance while some patients
prefer its tendency to cause less itching than chloroquine. Amodiaquine
is now available in a combined formulation with artesunate (ASAQ)
and is among the artemisinin-combination therapies recommended by the
World Health Organization. Combination with sulfadoxine=pyrimethamine is
not recommended.
The drug should be given in doses between 25 mg/kg and 35 mg/kg
over three days in a similar method to that used in chloroquine
administration. Adverse reactions are generally similar in severity and
type to that seen in chloroquine treatment. In addition, bradycardia, itching, nausea, vomiting and some abdominal pain have been recorded. Some blood and hepatic disorders have also been seen in a small number of patients.
Pyrimethamine
Pyrimethamine is used in the treatment of uncomplicated malaria. It is particularly useful in cases of chloroquine-resistant P. falciparum strains when combined with sulfadoxine. It acts by inhibiting dihydrofolate reductase in the parasite thus preventing the biosynthesis of purines and pyrimidines, thereby halting the processes of DNA replication, cell division
and reproduction. It acts primarily on the schizonts during the
erythrocytic phase, and nowadays is only used in concert with a sulfonamide.
Proguanil
Proguanil (chloroguanide) is a biguanide;
a synthetic derivative of pyrimidine. It was developed in 1945 by a
British Antimalarial research group. It has many mechanisms of action
but primarily is mediated through conversion to the active metabolitecycloguanil. This inhibits the malarial dihydrofolate reductase enzyme. Its most prominent effect is on the primary tissue stages of P. falciparum, P. vivax and P. ovale. It has no known effect against hypnozoites
therefore is not used in the prevention of relapse. It has a weak blood
schizonticidal activity and is not recommended for therapy of acute
infection. However it is useful in prophylaxis when combined with atovaquone or chloroquine
(in areas where there is no chloroquine resistance). 3 mg/kg is the
advised dosage per day, (hence approximate adult dosage is 200 mg). The
pharmacokinetic profile of the drugs indicates that a half dose, twice
daily maintains the plasma
levels with a greater level of consistency, thus giving a greater level
of protection. The proguanil- chloroquine combination does not provide
effective protection against resistant strains of P. falciparum.
There are very few side effects to proguanil, with slight hair loss and
mouth ulcers being occasionally reported following prophylactic use.
Proguanil hydrochloride is marketed as Paludrine by AstraZeneca.
Sulfonamides
Sulfadoxine and sulfamethoxypyridazine are specific inhibitors of the enzyme dihydropteroate synthetase in the tetrahydrofolate synthesis pathway of malaria parasites. They are structural analogs of p-aminobenzoic acid
(PABA) and compete with PABA to block its conversion to dihydrofolic
acid. Sulfonamides act on the schizont stages of the erythrocytic
(asexual) cycle. When administered alone sulfonamides are not
efficacious in treating malaria but co-administration with the
antifolate pyrimethamine, most commonly as fixed-dose sulfadoxine-pyrimethamine (Fansidar), produces synergistic effects sufficient to cure sensitive strains of malaria.
Sulfonamides are not recommended for chemoprophylaxis because of
rare but severe skin reactions experienced. However it is used
frequently for clinical episodes of the disease.
Mefloquine
Mefloquine was developed during the Vietnam War and is chemically related to quinine. It was developed to protect American troops against multi-drug resistantP. falciparum. It is a very potent blood schizonticide with a long half-life. It is thought to act by forming toxic heme complexes that damage parasitic food vacuoles. Mefloquine is effective in prophylaxis and for acute therapy. It is now used solely for the prevention of resistant strains of P. falciparum (usually combined with Artesunate) despite being effective against P. vivax, P. ovale and P. marlariae. Chloroquine/proguanil or sulfa drug-pyrimethamine combinations should be used in all other plasmodia infections.
The major commercial manufacturer of mefloquine-based malaria
treatment is Roche Pharmaceuticals, which markets the drug under the
trade name "Lariam". Lariam is fairly expensive at around three € per tablet (pricing of the year 2000).
A dose of 15–25 mg/kg is recommended, depending on the prevalence
of mefloquine resistance. The increased dosage is associated with a
much greater level of intolerance, most noticeably in young children;
with the drug inducing vomiting and esophagitis.
It was not recommended for use during the first trimester, although
considered safe during the second and third trimesters; nevertheless, in
October 2011, the Centers for Disease Control and Prevention (CDC)
changed its recommendation and approved use of Mefloquine for both
prophylaxis and treatment of malaria in all trimesters, after the Food
and Drug Administration (FDA) changed its categorization from C to B.
Mefloquine frequently produces side effects, including nausea, vomiting,
diarrhea, abdominal pain and dizziness. Several associations with
neurological events have been made, namely affective and anxiety disorders, hallucinations, sleep disturbances, psychosis, toxic encephalopathy, convulsions and delirium. Cardiovascular effects have been recorded with bradycardia and sinus arrhythmia being consistently recorded in 68% of patients treated with mefloquine (in one hospital-based study).
Mefloquine can only be taken for a period up to six months due to
side effects. After this, other drugs (such as those based on
paludrine/nivaquine) again need to be taken.
Atovaquone
Atovaquone is available in combination with proguanil under the name Malarone, albeit at a price higher than Lariam. It is commonly used in prophylaxis by travelers and used to treat falciparum malaria in developed countries.
A liquid oral suspension of Atovaquone is available under the name Mepron.
Primaquine
Primaquine is a highly active 8-aminoquinolone that is effective against P. falcipaum gametocytes but also acts on merozoites in the bloodstream and on hypnozoites, the dormant hepatic forms of P. vivax and P. ovale.
It is the only known drug to cure both relapsing malaria infections and
acute cases. The mechanism of action is not fully understood but it is
thought to block oxidative metabolism in Plasmodia. It can also be
combined with methylene blue.
For the prevention of relapse in P. vivax and P. ovale 0.15 mg/kg should be given for 14 days. As a gametocytocidal drug in P. falciparum
infections a single dose of 0.75 mg/kg repeated seven days later is
sufficient. This treatment method is only used in conjunction with
another effective blood schizonticidal drug. There are few significant
side effects although it has been shown that primaquine may cause anorexia, nausea, vomiting, cramps, chest weakness, anaemia, some suppression of myeloid activity and abdominal pains. In cases of over-dosage granulocytopenia may occur.
Artemisinin and derivatives
Artemisinin is a Chinese herb (qinghaosu) that has been used in the treatment of fevers for over 1,000 years, thus predating the use of Quinine in the western world. It is derived from the plant Artemisia annua, with the first documentation as a successful therapeutic agent in the treatment of malaria is in 340 AD by Ge Hong in his book Zhou Hou Bei Ji Fang (A Handbook of Prescriptions for Emergencies). Ge Hong extracted the artemesinin using a simple macerate, and this method is still in use today. The active compound was isolated first in 1971 and named artemisinin. It is a sesquiterpene lactone with a chemically rare peroxide bridge linkage. It
is thought to be responsible for the majority of its anti-malarial
action, although the target within the parasite remains controversial. At present it is strictly controlled under WHO guidelines as it has proven to be effective against all forms of multi-drug resistant P. falciparum,
thus every care is taken to ensure compliance and adherence together
with other behaviors associated with the development of resistance. It is also only given in combination with other anti-malarials.
Artemisinin
has a very rapid action and the vast majority of acute patients treated
show significant improvement within 1–3 days of receiving treatment. It has demonstrated the fastest clearance of all anti-malarials currently used and acts primarily on the trophozite phase, thus preventing progression of the disease.
Semi-synthetic artemisinin derivatives (e.g. artesunate, artemether)
are easier to use than the parent compound and are converted rapidly
once in the body to the active compound dihydroartemesinin.
On the first day of treatment 20 mg/kg is often given, and the dose
then reduced to 10 mg/kg per day for the six following days. Few side effects are associated with artemesinin use. However, headaches, nausea, vomiting, abnormal bleeding, dark urine, itching and some drug fever have been reported by a small number of patients. Some cardiac changes were reported during a clinical trial, notably non specific ST changes and a first degree atrioventricular block (these disappeared when the patients recovered from the malarial fever).
Artemether is a methylether
derivative of dihydroartemesinin. It is similar to artemesinin in mode
of action but demonstrates a reduced ability as a hypnozoiticidal
compound, instead acting more significantly to decrease gametocyte
carriage. Similar restrictions are in place, as with artemesinin, to
prevent the development of resistance, therefore it is only used in
combination therapy for severe acute cases of drug-resistant P. falciparum.
It should be administered in a 7-day course with 4 mg/kg given per day
for three days, followed by 1.6 mg/kg for three days. Side effects of
the drug are few but include potential neurotoxicity developing if high
doses are given.
Artesunate is a hemisuccinate derivative of the active metabolite dihydroartemisin. Currently
it is the most frequently used of all the artemesinin-type drugs. Its
only effect is mediated through a reduction in the gametocyte
transmission. It is used in combination therapy and is effective in
cases of uncomplicated P. falciparum. The dosage recommended by
the WHO is a five or seven day course (depending on the predicted
adherence level) of 4 mg/kg for three days (usually given in combination
with mefloquine) followed by 2 mg/kg for the remaining two or four
days. In large studies carried out on over 10,000 patients in Thailand
no adverse effects have been shown.
Dihydroartemisinin
is the active metabolite to which artemesinin is reduced. It is the
most effective artemesinin compound and the least stable. It has a
strong blood schizonticidal action and reduces gametocyte transmission.
It is used for therapeutic treatment of cases of resistant and
uncomplicated P. falciparum. 4 mg/kg doses are recommended on the
first day of therapy followed by 2 mg/kg for six days. As with
artesunate, no side effects to treatment have thus far been recorded.Arteether is an ethyl ether derivative of dihydroartemisinin. It is used in combination therapy for cases of uncomplicated resistant P. falciparum.
The recommended dosage is 150 mg/kg per day for three days given by IM
injections. With the exception of a small number of cases demonstrating
neurotoxicity following parenteral administration no side effects have been recorded.
Halofantrine
Halofantrine is a relatively new drug developed by the Walter Reed Army Institute of Research in the 1960s. It is a phenanthrene methanol, chemically related to Quinine and acts acting as a blood schizonticide effective against all Plasmodium parasites. Its mechanism of action is similar to other anti-malarials. Cytotoxic complexes are formed with ferritoporphyrin XI
that cause plasmodial membrane damage. Despite being effective against
drug resistant parasites, halofantrine is not commonly used in the
treatment (prophylactic or therapeutic) of malaria due to its high cost.
It has very variable bioavailability and has been shown to have
potentially high levels of cardiotoxicity.
It is still a useful drug and can be used in patients that are known to
be free of heart disease and are suffering from severe and resistant
forms of acute malaria. A popular drug based on halofantrine is Halfan.
The level of governmental control and the prescription-only basis on
which it can be used contributes to the cost, thus halofantrine is not
frequently used.
A dose of 8 mg/kg of halofantrine is advised to be given in three
doses at six-hour intervals for the duration of the clinical episode.
It is not recommended for children under 10 kg despite data supporting
the use and demonstrating that it is well tolerated. The most frequently
experienced side-effects include nausea, abdominal pain, diarrhea, and
itch. Severe ventricular dysrhythmias, occasionally causing death are seen when high doses are administered. This is due to prolongation of the QTc interval.
Halofantrine is not recommended for use in pregnancy and lactation, in
small children, or in patients that have taken mefloquine previously.
Lumefantrine
Lumefantrine is a relative of halofantrine that is used in some combination antimalarial regimens.
Doxycycline
Probably one of the more prevalent antimalarial drugs prescribed, due to its relative effectiveness and cheapness, doxycycline is a tetracycline compound derived from oxytetracycline.
The tetracyclines were one of the earliest groups of antibiotics to be
developed and are still used widely in many types of infection. It is a bacteriostatic agent that acts to inhibit the process of protein synthesis by binding to the 30Sribosomal subunit thus preventing the 50s and 30s units from bonding. Doxycycline is used primarily for chemoprophylaxis in areas where chloroquine resistance exists. It can also be used in combination with quinine to treat resistant cases of P. falciparum but has a very slow action in acute malaria, and should not be used as monotherapy.
When treating acute cases and given in combination with quinine;
100 mg of doxycycline should be given per day for seven days. In
prophylactic therapy, 100 mg (adult dose) of doxycycline should be given
every day during exposure to malaria.
The most commonly experienced side effects are permanent enamel hypoplasia, transient depression of bone growth, gastrointestinal disturbances and some increased levels of photosensitivity.
Due to its effect of bone and tooth growth it is not used in children
under 8, pregnant or lactating women and those with a known hepatic
dysfunction.
Tetracycline is only used in combination for the treatment of acute cases of P. falciparum
infections. This is due to its slow onset. Unlike doxycycline it is not
used in chemoprophylaxis. For tetracycline, 250 mg is the recommended
adult dosage (it should not be used in children) for five or seven days
depending on the level of adherence and compliance expected. Oesophageal
ulceration, gastrointestinal upset and interferences with the process
of ossification
and depression of bone growth are known to occur. The majority of side
effects associated with doxycycline are also experienced.
Clindamycin
Clindamycin is a derivative of lincomycin,
with a slow action against blood schizonticides. It is only used in
combination with quinine in the treatment of acute cases of resistant P. falciparum
infections and not as a prophylactic. Being more toxic than the other
antibiotic alternatives, it is used only in cases where the
Tetracyclines are contraindicated (for example in children).
Clindamycin should be given in conjunction with quinine as a
300 mg dose (in adults) four times a day for five days. The only side
effects recorded in patients taking clindamycin are nausea, vomiting and
abdominal pains and cramps. However these can be alleviated by
consuming large quantities of water and food when taking the drug. Pseudomembranous colitis (caused by Clostridium difficile) has also developed in some patients; this condition may be fatal in a small number of cases.
Resistance
Anti-malarial drug resistance
has been defined as: "the ability of a parasite to survive and/or
multiply despite the administration and absorption of a drug given in
doses equal to or higher than those usually recommended but within
tolerance of the subject. The drug in question must gain access to the
parasite or the infected red blood cell for the duration of the time
necessary for its normal action." Resistance to antimalarial drugs is common.
In most instances this refers to parasites that remain following on from
an observed treatment; thus, it excludes all cases where anti-malarial
prophylaxis has failed.
In order for a case to be defined as resistant, the patient in question
must have received a known and observed anti-malarial therapy while the
blood drug and metabolite concentrations are monitored concurrently;
techniques used to demonstrate this include in vivo, in vitro, and animal model testing, and more recently developed molecular techniques.
Drug resistant parasites are often used to explain malaria
treatment failure. However, they are two potentially very different
clinical scenarios. The failure to clear parasitemia
and recover from an acute clinical episode when a suitable treatment
has been given is anti-malarial resistance in its true form. Drug
resistance may lead to treatment failure, but treatment failure is not
necessarily caused by drug resistance despite assisting with its
development. A multitude of factors can be involved in the processes
including problems with non-compliance and adherence, poor drug quality,
interactions with other pharmaceuticals, poor absorption, misdiagnosis
and incorrect doses being given. The majority of these factors also
contribute to the development of drug resistance.
The generation of resistance can be complicated and varies between Plasmodium species. It is generally accepted to be initiated primarily through a spontaneous mutation that provides some evolutionary benefit, thus giving the anti-malarial used a reduced level of sensitivity. This can be caused by a single point mutation
or multiple mutations. In most instances a mutation will be fatal for
the parasite or the drug pressure will remove parasites that remain
susceptible, however some resistant parasites will survive. Resistance
can become firmly established within a parasite population, existing for
long periods of time.
The first type of resistance to be acknowledged was to
chloroquine in Thailand in 1957. The biological mechanism behind this
resistance was subsequently discovered to be related to the development
of an efflux mechanism that expels chloroquine from the parasite before
the level required to effectively inhibit the process of haem
polymerization (that is necessary to prevent buildup of the toxic
byproducts formed by haemoglobin digestion). This theory has been
supported by evidence showing that resistance can be effectively
reversed on the addition of substances which halt the efflux. The
resistance of other quinolone anti-malarials such as amiodiaquine,
mefloquine, halofantrine and quinine are thought to have occurred by
similar mechanisms.
Plasmodium have developed resistance against antifolate
combination drugs, the most commonly used being sulfadoxine and
pyrimethamine. Two gene mutations are thought to be responsible,
allowing synergistic blockages of two enzymes involved in folate synthesis. Regional variations of specific mutations give differing levels of resistance.
Atovaquone
is recommended to be used only in combination with another
anti-malarial compound as the selection of resistant parasites occurs
very quickly when used in mono-therapy. Resistance is thought to
originate from a single-point mutation in the gene coding for
cytochrome-b.
Spread of resistance
There is no single factor that confers the greatest degree of
influence on the spread of drug resistance, but a number of plausible
causes associated with an increase have been acknowledged. These include
aspects of economics, human behaviour, pharmacokinetics, and the
biology of vectors and parasites.
The most influential causes are examined below:
The biological influences are based on the parasites ability to
survive the presence of an anti-malarial thus enabling the persistence
of resistance and the potential for further transmission despite
treatment. In normal circumstances any parasites that persist after
treatment are destroyed by the host's immune system, therefore any
factors that act to reduce the elimination of parasites could facilitate
the development of resistance. This attempts to explain the poorer
response associated with immunocompromised individuals, pregnant women and young children.
There has been evidence to suggest that certain parasite-vector
combinations can alternatively enhance or inhibit the transmission of
resistant parasites, causing 'pocket-like' areas of resistance.
The use of anti-malarials developed from similar basic chemical
compounds can increase the rate of resistance development, for example
cross-resistance to chloroquine and amiodiaquine, two 4-aminoquinolones
and mefloquine conferring resistance to quinine and halofantrine. This
phenomenon may reduce the usefulness of newly developed therapies prior
to large-scale usage.
The resistance to anti-malarials may be increased by a process found in some species of Plasmodium, where a degree of phenotypic plasticity
was exhibited, allowing the rapid development of resistance to a new
drug, even if the drug has not been previously experienced.
The pharmacokinetics of the chosen anti-malarial are key; the
decision of choosing a long half-life over a drug that is metabolised
quickly is complex and still remains unclear. Drugs with shorter
half-life's require more frequent administration to maintain the correct
plasma concentrations, therefore potentially presenting more problems
if levels of adherence and compliance are unreliable, but longer-lasting
drugs can increase the development of resistance due to prolonged
periods of low drug concentration.
The pharmacokinetics of anti-malarials is important when using
combination therapy. Mismatched drug combinations, for example having an
'unprotected' period where one drug dominates can seriously increase
the likelihood of selection for resistant parasites.
Ecologically there is a linkage between the level of transmission
and the development of resistance, however at present this still remains
unclear.
The treatment regime prescribed can have a substantial influence on
the development of resistance. This can involve the drug intake,
combination and interactions as well as the drug's pharmacokinetic and
dynamic properties.
Prevention
The prevention of anti-malarial drug resistance is of enormous public health importance. It can be assumed that no therapy currently
under development or to be developed in the foreseeable future will be
totally protective against malaria. In accordance with this, there is
the possibility of resistance developing to any given therapy that is
developed. This is a serious concern, as the rate at which new drugs are
produced by no means matches the rate of the development of resistance.
In addition, the most newly developed therapeutics tend to be the most
expensive and are required in the largest quantities by some of the
poorest areas of the world. Therefore, it is apparent that the degree to
which malaria can be controlled depends on the careful use of the
existing drugs to limit, insofar as it is possible, any further
development of resistance.
Provisions essential to this process include the delivery of fast
primary care where staff are well trained and supported with the
necessary supplies for efficient treatment. This in itself is inadequate
in large areas where malaria is endemic thus presenting an initial
problem. One method proposed that aims to avoid the fundamental lack in
certain countries' health care infrastructure
is the privatisation of some areas, thus enabling drugs to be purchased
on the open market from sources that are not officially related to the
health care industry. Although this is now gaining some support there
are many problems related to limited access and improper drug use, which
could potentially increase the rate of resistance development to an
even greater extent.
There are two general approaches to preventing the spread of resistance: preventing malaria infections, and preventing the transmission of resistant parasites.
Preventing malaria infections developing has a substantial effect
on the potential rate of development of resistance, by directly
reducing the number of cases of malaria thus decreasing the need for
anti-malarial therapy.
Preventing the transmission of resistant parasites limits the risk of
resistant malarial infections becoming endemic and can be controlled by a
variety of non-medical methods including insecticide-treated bed nets, indoor residual spraying, environmental controls (such as swamp draining) and personal protective methods such as using mosquito repellent.
Chemoprophylaxis is also important in the transmission of malaria
infection and resistance in defined populations (for example travelers).
A hope for future of anti-malarial therapy is the development of an effective malaria vaccine.
This could have enormous public health benefits, providing a
cost-effective and easily applicable approach to preventing not only the
onset of malaria but the transmission of gametocytes, thus reducing the
risk of resistance developing. Anti-malarial therapy also could be
diversified by combining a potentially effective vaccine with current chemotherapy, thereby reducing the chance of vaccine resistance developing.
Combination therapy
The problem of the development of malaria resistance must be weighed
against the essential goal of anti-malarial care; that is to reduce morbidity
and mortality. Thus a balance must be reached that attempts to achieve
both goals while not compromising either too much by doing so. The most
successful attempts so far have been in the administration of
combination therapy. This can be defined as, 'the simultaneous use of
two or more blood schizonticidal drugs with independent modes of action
and different biochemical targets in the parasite'.
There is much evidence to support the use of combination therapies,
some of which has been discussed previously, however several problems
prevent the wide use in the areas where its use is most advisable. These
include: problems identifying the most suitable drug for different
epidemiological situations, the expense of combined therapy (it is over
10 times more expensive than traditional mono-therapy), how soon the
programmes should be introduced and problems linked with policy
implementation and issues of compliance.
The combinations of drugs currently
prescribed can be divided into two categories: non-artemesinin-based
combinations and artemesinin based combinations. It is also important to
distinguish fixed-dose combination therapies (in which two or
more drugs are co-formulated into a single tablet) from combinations
achieved by taking two separate antimalarials.
Non-artemisinin based combinations
Components
Description
Dose
Sulfadoxine-pyrimethamine (SP) (Fansidar)
This fixed-dose combination has been used for many years, causes few
adverse effects, is cheap and effective in a single dose, thus
decreasing problems associated with adherence and compliance. In
technical terms Fansidar is not generally considered a true combination
therapy since the components do not possess independent curative
activity. Fansidar should no longer be used alone for treatment of falciparum malaria.
25 mg/kg of sulfadoxine and 1.25 mg/kg of pyrimethamine.
SP plus chloroquine
High levels of resistance to one or both components means this
combination is effective in few locations and it is not recommended by
the World Health Organization (WHO).
Chloroquine 25 mg/kg over three days with a single dose of SP as described above.
SP plus amodiaquine
This combination has been shown to produce a faster rate of clinical
recovery than SP and chloroquine, but is clearly inferior to
artemisinin-based combinations (ACTs) for the treatment of malaria.
10 mg/kg of Amodiaquine per day for three days with a single standard dose of SP.
SP plus mefloquine (Fansimef)
This single dose pill offered obvious advantages of convenience over
more complex regimes but it has not been recommended for use for many
years owing to widespread resistance to the components.
Quinine plus tetracycline/doxycycline
This combination retains a high cure rate in many areas. Problems
with this regime include the relatively complicated drug regimen, where
quinine must be taken every eight hours for seven days. Additionally,
there are significant side effects with quinine ('cinchonism')
and tetracyclines are contraindicated in children and pregnant women
(these groups should use clindamycin instead). With the advent of
artemisinin-combination therapies, quinine-based treatment is less
popular than previously.
Quinine 10 mg/kg doses every eight hours and tetracycline in 4 mg/kg doses every six hours for seven days.
Artemisinin-based combination therapies should be used in preference
to amodiaquine plus sulfadoxine-pyrimethamine for the treatment of
uncomplicated P. falciparum malaria.
Artemisinin-based combination therapies (ACTs)
Artemesinin
has a very different mode of action than conventional anti-malarials
(see information above), which makes it particularly useful in the
treatment of resistant infections. However, to prevent the development
of resistance to this drug it is only recommended in combination with
another non-artemesinin based therapy. It produces a very rapid
reduction in the parasite biomass with an associated reduction in
clinical symptoms and is known to cause a reduction in the transmission
of gametocytes thus decreasing the potential for the spread of resistant
alleles. At present there is no known resistance to Artemesinin (though
some resistant strains may be emerging) and very few reported side-effects to drug usage, however this data is limited.
This combination has been tested and proved to be efficacious in
many areas where amodiaquine retains some efficacy. A potential
disadvantage is a suggested link with neutropenia. It's recommended by the WHO for uncomplicated falciparum malaria.
Dosage is as a fixed-dose combination (ASAQ) recommended as 4 mg/kg of Artesunate and 10 mg/kg of Amodiaquine per day for three days.
This has been used as an efficacious first-line treatment regimen in
areas of Thailand for many years. Mefloquine is known to cause vomiting
in children and induces some neuropsychiatric and cardiotoxic effects.
These adverse reactions seem to be reduced when the drug is combined
with artesunate, it is suggested that this is due to a delayed onset of
action of mefloquine. This is not considered a viable option to be
introduced in Africa due to the long half-life of mefloquine, which
potentially could exert a high selection pressure on parasites. It's
recommended by the WHO for uncomplicated falciparum malaria.
The standard dose required is 4 mg/kg per day of Artesunate plus
25 mg/kg of Mefloquine as a split dose of 15 mg/kg on day two and
10 mg/kg on day three.
This combination has been extensively tested in 16 clinical trials,
proving effective in children under five and has been shown to be better
tolerated than artesunate plus mefloquine combinations. There are no
serious side effects documented but the drug is not recommended in
pregnant or lactating women due to limited safety testing in these
groups. This is the most viable option for widespread use and is
available in fixed-dose formulas thus increasing compliance and
adherence. It's recommended by the WHO for uncomplicated falciparum malaria.
This is a well tolerated combination but the overall level of
efficacy still depends on the level of resistance to sulfadoxine and
pyrimethamine thus limiting is usage. It is recommended by the WHO for
uncomplicated falciparum malaria.
It is recommended in doses of 4 mg/kg of Artesunate per day for three days and a single dose of 25 mg/kg of SP.
Has been studied mainly in China, Vietnam and other countries in
SEAsia. The drug has been shown to be highly efficacious (greater than
90%). It's recommended by the WHO for uncomplicated falciparum malaria.
Artesinin/piperaguine/primaquine (Fast Elimination of Malaria through Source Eradication (FEMSE))
This protocol involves three doses of Artequick, spaced a month apart. The first dose is accompanied by one of primaquine.
An experimental program in the Comoros islands employed the protocol.
At the outset, more than 90% of the inhabitants of some villages had
malaria. On one island the number of cases fell by 95%. In 2012, on the
second island, the number of cases fell by 97%.
Pyramax developed by Shin Poong Pharmaceutical and Medicines for
Malaria Venture (MMV). This is a first fixed-dose artemisinin-based
combination therapy to be granted a positive scientific opinion for
efficacy, safety and quality from European Medicines Agency (EMA) under
Article 58 for the treatment of P. falciparum and P. vivax
in adults and children over 20 kg based on five multi-centre phase III
trials conducted in Africa and South-East Asia. Pyramax has been shown
to be highly efficacious (greater than 97%) in both species and only ACT
approved by stringent regulatory authority for treatment of both P. falciparum and P vivax by now.
Other combinations
Several other anti-malarial combinations have been used or are in development. For example, Chlorproguanil-dapsone and artesunate (CDA) appears efficacious but the problem of haemolysis in patients with glucose-6-phosphate dehydrogenase (G6PD) deficiency is likely to prevent widespread use.
By type of malaria
Antimalarial drugs and combinations may also be sorted according to the type of malaria in which they are used.
Falciparum malaria
Artemisinin-based combination therapies (ACTs) are the recommended antimalarial treatments for uncomplicated malaria caused by P. falciparum. The choice of ACT in a country or region will be based on the level of resistance to the constituents in the combination. For pregnant women, the recommended first-line treatment during the first trimester is quinine plus clindamycin to be given for seven days.
In second and third trimesters, it is recommended to give ACTs known to
be effective in the country/region or artesunate plus clindamycin for
seven days, or quinine plus clindamycin to be given for seven days. Lactating women should receive standard antimalarial treatment (including ACTs) except for dapsone, primaquine and tetracyclines.
In infants and young children, it is recommended to give ACTs for
first-line treatment, with attention to accurate dosing and ensuring the
administered dose is retained.
In severe falciparum malaria, it is recommended that rapid
clinical assessment and confirmation of the diagnosis is made, followed
by administration of full doses of parenteral antimalarial treatment
without delay with whichever effective antimalarial is first available. For adults, intravenous (IV) or intramuscular (IM) artesunate is recommended. Quinine is an acceptable alternative if parenteral artesunate is not available.
Parenteral antimalarials should be administered for a minimum of 24 h
in the treatment of severe malaria, irrespective of the patient's
ability to tolerate oral medication earlier. Thereafter, it is recommended to complete treatment by giving a complete course of any of the following:
an ACT
artesunate plus clindamycin or doxycycline;
quinine plus clindamycin or doxycycline.
Vivax malaria
Chloroquine remains the treatment of choice for vivax malaria, except in Indonesia's Irian Jaya (Western New Guinea) region and the geographically contiguous Papua New Guinea, where chloroquine resistance is common (up to 20% resistance).


.png)