Nootropics (/noʊ.əˈtrɒpɪks/noh-ə-TROP-iks) (colloquial: smart drugs and cognitive enhancers) are drugs, supplements, and other substances that may improve cognitive function, particularly executive functions, memory, creativity, or motivation, in healthy individuals.
While many substances are purported to improve cognition, research is
at a preliminary stage as of 2018, and the effects of the majority of
these agents are not fully determined.
The use of cognition-enhancing drugs by healthy individuals in the absence of a medical indication spans numerous controversial issues, including the ethics and fairness of their use, concerns over adverse effects, and the diversion of prescription drugs for nonmedical uses, among others. Nonetheless, the international sales of cognition-enhancing supplements exceeded US$1 billion in 2015 when global demand for these compounds grew.
The word nootropic was coined in 1972 by a Romanian psychologist and chemist, Corneliu E. Giurgea, from the Greek words νοῦς (nous), or "mind", and τρέπειν (trepein), meaning to bend or turn.
Availability and prevalence
In 2008, the most commonly used class of drug was stimulants, such as caffeine. Manufacturer's marketing claims for dietary supplements are usually not formally tested and verified by independent entities.
Use by students
The use of prescription stimulants is especially prevalent among students. Surveys suggest that 0.7–4.5% of German students have used cognitive enhancers in their lifetime. Stimulants such as dimethylamylamine and methylphenidate are used on college campuses and by younger groups. Based upon studies of self-reported illicit stimulant use, 5–35% of college students use diverted ADHD stimulants, which are primarily intended for performance enhancement rather than as recreational drugs.
Several factors positively and negatively influence an individual's
willingness to use a drug for the purpose of enhancing cognitive
performance. Among them are personal characteristics, drug
characteristics, and characteristics of the social context.
Side effects
The main concern with pharmaceutical drugs is adverse effects, which also apply to nootropics with undefined effects. Long-term safety evidence is typically unavailable for nootropics. Racetams — piracetam and other compounds that are structurally related to piracetam — have few serious adverse effects and low toxicity, but there is little evidence that they enhance cognition in people having no cognitive impairments.
In the United States, dietary supplements may be marketed if the manufacturer can show that the supplement is generally recognized as safe,
and if the manufacturer does not make any claims about using the
supplement to treat or prevent any disease or condition; supplements
that contain drugs or advertise health claims are illegal under US law.
Amphetamine – systematic reviews and meta-analyses report that low-dose amphetamine improved cognitive functions (e.g., inhibitory control, episodic memory, working memory, and aspects of attention) in healthy people and in individuals with ADHD. A 2014 systematic review noted that low doses of amphetamine also improved memory consolidation, in turn leading to improved recall of information in non-ADHD youth. It also improves task saliency (motivation to perform a task) and performance on tedious tasks that required a high degree of effort.
Eugeroics (armodafinil and modafinil) – are classified as "wakefulness promoting" agents; modafinil increased alertness, particularly in sleep deprived individuals, and was noted to facilitate reasoning and problem solving in non-ADHD youth.
In a systematic review of small, preliminary studies where the effects
of modafinil were examined, when simple psychometric assessments were
considered, modafinil intake appeared to enhance executive function. Modafinil does not produce improvements in mood or motivation in sleep deprived or non-sleep deprived individuals.
Caffeine – a meta-analysis found an increase in alertness and attentional performance.
Nicotine – a meta-analysis of 41 clinical studies concluded that nicotine
or smoking caused improvements in alerting and orienting attention and
episodic and working memory and slightly improved fine motor
performance.
According to the US Food and Drug Administration,
"Piracetam is not a vitamin, mineral, amino acid, herb or other
botanical, or dietary substance for use by man to supplement the diet by
increasing the total dietary intake. Further, piracetam is not a
concentrate, metabolite, constituent, extract or combination of any such
dietary ingredient. [...] Accordingly, these products are drugs, under
section 201(g)(1)(C) of the Act, 21 U.S.C. § 321(g)(1)(C), because they
are not foods and they are intended to affect the structure or any
function of the body. Moreover, these products are new drugs as defined
by section 201(p) of the Act, 21 U.S.C. § 321(p), because they are not
generally recognized as safe and effective for use under the conditions
prescribed, recommended, or suggested in their labeling."
Miscellaneous
L-Theanine – A 2014 systematic review and meta-analysis found that concurrent caffeine and L-theanine use had synergistic psychoactive effects that promoted alertness, attention, and task switching; these effects were most pronounced during the first hour post-dose. However, the European Food Safety Authority
reported that, when L-theanine is used by itself (i.e. without
caffeine), there is insufficient information to determine if these
effects exist.
Panax ginseng – A review by the Cochrane Collaboration concluded that "there is a lack of convincing evidence to show a cognitive enhancing effect of Panax ginseng in healthy participants and no high quality evidence about its efficacy in patients with dementia." According to the National Center for Complementary and Integrative Health,
"[a]lthough Asian ginseng has been widely studied for a variety of
uses, research results to date do not conclusively support health claims
associated with the herb."
Ginkgo biloba – An extract of Ginkgo biloba leaf is marketed in dietary supplement form with claims it can enhance cognitive function
in people without known cognitive problems, although there is no
high-quality evidence to support such effects on memory or attention in
healthy people.
Salvia officinalis (sage) – Some research has suggested certain extracts of Salvia officinalis
may have positive effects on human brain function, but due to
significant methodological problems, no firm conclusions can be drawn. The thujone present in Salvia extracts may be neurotoxic.
Null findings in systematic reviews
Omega-3 fatty acids: DHA and EPA – two Cochrane Collaboration
reviews on the use of supplemental omega-3 fatty acids for ADHD and
learning disorders conclude that there is limited evidence of treatment
benefits for either disorder. Two other systematic reviews noted no cognition-enhancing effects in the general population or middle-aged and older adults.
Folate – no cognition-enhancing effects in middle-aged and older adults.
Vitamin B6 – no cognition-enhancing effects in middle-aged and older adults.
Vitamin B12 – no cognition-enhancing effects in middle-aged and older adults.
Vitamin E – no cognition-enhancing effects in middle-aged and older adults.
Pramipexole – no significant cognition-enhancing effects in healthy individuals.
Guanfacine – no significant cognition-enhancing effects in healthy individuals.
Clonidine – no significant cognition-enhancing effects in healthy individuals.
Fexofenadine – no significant cognition-enhancing effects in healthy individuals.
The history of aspirin (IUPAC name acetylsalicylic acid ) begins with its synthesis and manufacture in 1899. Before that, salicylic acid had been used medicinally since antiquity. Medicines made from willow and other salicylate-rich plants appear in clay tablets from ancient Sumer as well as the Ebers Papyrus from ancient Egypt.
Hippocrates referred to their use of salicylic tea to reduce fevers
around 400 BC, and were part of the pharmacopoeia of Western medicine in
classical antiquity and the Middle Ages. Willow bark extract became recognized for its specific effects on fever, pain and inflammation in the mid-eighteenth century. By the nineteenth century pharmacists were experimenting with and prescribing a variety of chemicals related to salicylic acid, the active component of willow extract.
In 1853, chemist Charles Frédéric Gerhardt treated acetyl chloride with sodium salicylate to produce acetylsalicylic acid for the first time;
in the second half of the nineteenth century, other academic chemists
established the compound's chemical structure and devised more efficient
methods of synthesis. In 1897, scientists at the drug and dye firm Bayer
began investigating acetylsalicylic acid as a less-irritating
replacement for standard common salicylate medicines, and identified a
new way to synthesize it. By 1899, Bayer had dubbed this drug Aspirin and was selling it around the world. The word Aspirin
was Bayer's brand name, rather than the generic name of the drug;
however, Bayer's rights to the trademark were lost or sold in many
countries. Aspirin's popularity grew over the first half of the
twentieth century leading to fierce competition with the proliferation
of aspirin brands and products.
Aspirin's popularity declined after the development of acetaminophen/paracetamol in 1956 and ibuprofen in 1962. In the 1960s and 1970s, John Vane and others discovered the basic mechanism of aspirin's effects,
while clinical trials and other studies from the 1960s to the 1980s
established aspirin's efficacy as an anti-clotting agent that reduces
the risk of clotting diseases.
Aspirin sales revived considerably in the last decades of the twentieth
century, and remain strong in the twenty-first with widespread use as a
preventive treatment for heart attacks and strokes.
Early history of salicylates
Medicines derived from willow trees and other salicylate-rich plants have been part of pharmacopoeias at least dating back to ancient Sumer. The Ebers Papyrus, an Egyptian medical text from ca. 1543 BCE, mentions use of willow and myrtle (another salicylate-rich plant) to treat fever and pain.
Willow bark preparations became a standard part of the materia medica of Western medicine beginning at least with the Greek physician Hippocrates
in the fifth century BCE; he recommended chewing on willow bark to
relieve pain or fever, and drinking tea made from it to relieve pain
during childbirth. The Roman encyclopedist Celsus, in his De Medicina of ca. 30 AD, suggested willow leaf extract to treat the four signs of inflammation: redness, heat, swelling and pain. Willow treatments also appeared in Dioscorides's De Materia Medica, and Pliny the Elder's Natural History. By the time of Galen, willow bark was commonly used throughout the Roman and Arab worlds, as a small part of a large, growing botanical pharmacopoeia.
18th and 19th centuries
Edward Stone found that the bark of the white willow (Salix alba) could substitute for Peruvian bark in the treatment of ague.
The major turning point for salicylate medicines came in 1763, when a letter from English chaplain Edward Stone was read at a meeting of the Royal Society,
describing the dramatic power of willow bark extract to cure ague—an
ill-defined constellation of symptoms, including intermittent fever,
pain, and fatigue, that primarily referred to malaria. Inspired by the doctrine of signatures
to search for a treatment for agues near the brackish waters that were
known to cause it, Stone had tasted the bark of a willow tree in 1758
and noticed an astringency reminiscent of the standard—and expensive—ague cure of Peruvian bark.
He collected, dried, and powdered a substantial amount of willow bark,
and over the next five years tested it on a number of people sick with
fever and agues. In his letter, Stone reported consistent success,
describing willow extract's effects as identical to Peruvian bark,
though a little less potent. (In fact, the active ingredient of Peruvian bark was quinine, which attacked the infectious cause of malaria, while the active ingredient of willow extract, salicin, relieved the symptoms of malaria but could not cure it.) Stone's letter (mistakenly attributed to Edmund rather than Edward Stone) was printed in Philosophical Transactions, and by the end of the 18th century willow was gaining popularity as an inexpensive substitute for Peruvian bark.
In the 19th century, as the young discipline of organic chemistry
began to grow in Europe, scientists attempted to isolate and purify the
active components of many medicines, including willow bark. After
unsuccessful attempts by Italian chemists Brugnatelli and Fontana in
1826, Joseph Buchner obtained relatively pure salicin crystals in 1828; the following year, Henri Leroux developed another procedure for extracting modest yields of salicin.
In 1834, Swiss pharmacist Johann Pagenstecher discovered what he
thought was a new pain-reducing substance, isolated from the common
remedy of meadowsweet (Spiraea ulmaria). By 1838, Italian chemist Raffaele Piria found a method of obtaining a more potent acid form of willow extract, which he named salicylic acid. The German chemist who had been working to identify the Spiraea extract, Karl Jacob Löwig, soon realized that it was in fact the same salicylic acid that Piria had found.
Meadowsweet (Filipendula ulmaria). Tea made from its flowers are an old folk remedy against fever and pain.
Salicylate medicines—including salicin, salicylic acid, and sodium
salicylate— were difficult and wasteful to extract from plants, and in
1860 Hermann Kolbe worked out a way to synthesize salicylic acid.
Throughout the late 1800s use of salicylates grew considerably, and
physicians increasingly knew what to expect from these medicines:
reduction of pain, fever, and inflammation. However, the unpleasant side effects, particularly gastric irritation, limited their usefulness, as did their intense bitterness. By the 1880s, the German chemical industry, jump-started by the lucrative development of dyes from coal tar, was branching out to investigate the potential of new tar-derived medicines. The turning point was the advent of Kalle & Company's Antifebrine,
the branded version of the well-known dye derivative acetanilide—the
fever-reducing properties of which were discovered by accident in 1886.
Antifebrine's success inspired Carl Duisberg, the head of research at the small dye firm Friedrich Bayer & Company, to start a systematic search for other chemical fever-reducers. Bayer chemists soon developed Phenacetin, followed by the sedativesSulfonal and Trional.
Synthesis of acetylsalicylic acid
Upon
taking control of Bayer's overall management in 1890, Duisberg began to
expand the company's drug research program. He created a pharmaceutical
group for creating new drugs, headed by former university chemist Arthur Eichengrün, and a pharmacology group for testing the drugs, headed by Heinrich Dreser (beginning in 1897, after periods under Wilhelm Siebel and Hermann Hildebrandt). In 1894, the young chemist Felix Hoffmann
joined the pharmaceutical group. Dreser, Eichengrün and Hoffmann would
be the key figures in the development of acetylsalicylic acid as the
drug Aspirin (though their respective roles have been the subject of
some contention).
In 1897, Hoffmann started working to find a less irritating
substitute for salicylic acid. It is generally accepted that he turned
to this idea because his father was suffering the side effects of taking
sodium salicylate for rheumatism.
In 1853, Charles Frédéric Gerhardt had published the first methods to prepare acetylsalicylic acid. In the course of his work on the synthesis and properties of various acid anhydrides, he mixed acetyl chloride with a sodium salt of salicylic acid (sodium salicylate). A vigorous reaction ensued, and the resulting melt soon solidified. Since no structural theory existed at that time Gerhardt called the compound he obtained "salicylic-acetic anhydride" (wasserfreie Salicylsäure-Essigsäure). When Gerhardt tried to dissolve the solid in a diluted solution of sodium carbonate it immediately decomposed to sodium salts of salicylic and acetic acids. In 1859 an Austrian chemist, Hugo von Gilm, obtained analytically pure acetylsalicylic acid (which he called acetylierte Salicylsäure, acetylated salicylic acid) by a reaction of salicylic acid and acetyl chloride.
In 1869 Schröder, Prinzhorn and Kraut repeated both Gerhardt's (from
sodium salicylate) and von Gilm's (from salicylic acid) syntheses and
concluded that both reactions gave the same compound—acetylsalicylic
acid. (Prinzhorn is credited in the paper with conducting the
experiments.) They were first to assign to it the correct structure with
the acetyl group connected to the phenolic oxygen.
It is likely that Hoffmann did as most chemists have always done,
starting by studying the literature and recreating the published
methods. On 10 August 1897 (according to his laboratory notebooks), Hoffmann found a better method for making ASA, from salicylic acid refluxed with acetic anhydride.
Eichengrün sent ASA to Dreser's pharmacology group for testing,
and the initial results were very positive. The next step would normally
have been clinical trials,
but Dreser opposed further investigation of ASA because of salicylic
acid's reputation for weakening the heart—possibly a side effect of the
high doses often used to treat rheumatism. Dreser's group was soon busy testing Felix Hoffmann's next chemical success: diacetylmorphine (which the Bayer team soon branded as heroin
because of the heroic feeling it gave them). Eichengrün, frustrated by
Dreser's rejection of ASA, went directly to Bayer's Berlin
representative Felix Goldmann to arrange low-profile trials with
doctors. Though the results of those trials were also very positive,
with no reports of the typical salicylic acid complications, Dreser
still demurred. However, Carl Duisberg intervened and scheduled full
testing. Soon, Dreser admitted ASA's potential and Bayer decided to
proceed with production. Dreser wrote a report of the findings to
publicize the new drug; in it, he omitted any mention of Hoffmann or
Eichengrün.
He would also be the only one of the three to receive royalties for the
drug (for testing it), since it was ineligible for any patent the
chemists might have taken out for creating it. For many years, however,
he attributed Aspirin's discovery solely to Hoffmann.
The controversy over who was primarily responsible for aspirin's
development spread through much of the twentieth century and into the
twenty-first. Although aspirin's origin was in academic research and
Bayer was not the first to synthesize it, as of 2016 Bayer still
described Hoffman as having "discovered a pain-relieving, fever-lowering
and anti-inflammatory substance."
Historians and others have also challenged Bayer's early accounts of
Bayer's synthesis, in which Hoffmann was primarily responsible for the
Bayer breakthrough. In 1949, shortly before his death, Eichengrün wrote
an article, "Fifty Years of Asprin", claiming that he had not told
Hoffmann the purpose of his research, meaning that Hoffmann merely
carried out Eichengrün's research plan, and that the drug would never
have gone to the market without his direction. This claim was later
supported by research conducted by historian Walter Sneader.
Axel Helmstaedter, General Secretary of the International Society for
the History of Pharmacy, subsequently questioned the novelty of
Sneader’s research, noting that several earlier articles discussed the
Hoffmann–Eichengrün controversy in detail.
Bayer countered Sneader in a press release stating that according to
the records, Hoffmann and Eichengrün held equal positions, and
Eichengrün was not Hoffmann's supervisor. Hoffmann was named on the US
Patent as the inventor, which Sneader did not mention. Eichengrün, who
left Bayer in 1908, had multiple opportunities to claim the priority and
had never before 1949 done it; he neither claimed nor received any
percentage of the profit from aspirin sales.
Naming the drug
Spirea, or meadowsweet, is the German namesake of Spirsäure (salicylic acid), and ultimately aspirin.
The name Aspirin was derived from the name of the chemical ASA—Acetylspirsäure in German. Spirsäure (salicylic acid) was named for the meadowsweet plant, Spirea ulmaria, from which it could be derived.Aspirin took a- for the acetylation, -spir- from Spirsäure, and added -in
as a typical drug name ending to make it easy to say. In the final
round of naming proposals that circulated through Bayer, it came down to
Aspirin and Euspirin; Aspirin, they feared, might remind customers of aspiration, but Arthur Eichengrün argued that Eu-
(meaning "good") was inappropriate because it usually indicated an
improvement over an earlier version of a similar drug. Since the
substance itself was already known, Bayer intended to use the new name
to establish their drug as something new; in January 1899 they settled
on Aspirin.
Rights and sale
Under Carl Duisberg's leadership, Bayer was firmly committed to the standards of ethical drugs, as opposed to patent medicines. Ethical drugs were drugs that could be obtained only through a pharmacist, usually with a doctor's prescription. Advertising drugs directly to consumers
was considered unethical and strongly opposed by many medical
organizations; that was the domain of patent medicines. Therefore, Bayer
was limited to marketing Aspirin directly to doctors.
When production of Aspirin began in 1899, Bayer sent out small
packets of the drug to doctors, pharmacists and hospitals, advising them
of Aspirin's uses and encouraging them to publish about the drug's
effects and effectiveness. As positive results came in and enthusiasm
grew, Bayer sought to secure patent and trademark wherever possible. It
was ineligible for patent in Germany (despite being accepted briefly
before the decision was overturned), but Aspirin was patented in Britain
(filed 22 December 1898) and the United States (US Patent 644,077
issued 27 February 1900). The British patent was overturned in 1905, the
American patent was also besieged but was ultimately upheld.
Faced with growing legal and illegal competition for the globally
marketed ASA, Bayer worked to cement the connection between Bayer and
Aspirin. One strategy it developed was to switch from distributing
Aspirin powder for pharmacists to press into pill form to distributing
standardized tablets—complete with the distinctive Bayer cross logo. In 1903 the company set up an American subsidiary, with a converted factory in Rensselaer, New York, to produce Aspirin for the American market without paying import duties.
Bayer also sued the most egregious patent violators and smugglers. The
company's attempts to hold onto its Aspirin sales incited criticism from
muckraking journalists and the American Medical Association, especially after the 1906 Pure Food and Drug Act that prevented trademarked drugs from being listed in the United States Pharmacopeia;
Bayer listed ASA with an intentionally convoluted generic name
(monoacetic acid ester of salicylic acid) to discourage doctors
referring to anything but Aspirin.
World War I and Bayer
By the outbreak of World War I
in 1914, Bayer was facing competition in all its major markets from
local ASA producers as well as other German drug firms (particularly
Heyden and Hoechst). The British market was immediately closed to the German companies, but British manufacturing could not meet the demand—especially with phenol
supplies, necessary for ASA synthesis, largely being used for
explosives manufacture. On 5 February 1915, Bayer's UK trademarks were
voided, so that any company could use the term aspirin. The Australian market was taken over by Aspro, after the makers of Nicholas-Aspirin lost a short-lived exclusive right to the aspirin
name there. In the United States, Bayer was still under German
control—though the war disrupted the links between the American Bayer
plant and the German Bayer headquarters—but phenol shortage threatened
to reduce aspirin production to a trickle, and imports across the Atlantic Ocean were blocked by the Royal Navy.
To secure phenol for aspirin production, and at the same time
indirectly aid the German war effort, German agents in the United States
orchestrated what became known as the Great Phenol Plot.
By 1915, the price of phenol rose to the point that Bayer's aspirin
plant was forced to drastically cut production. This was especially
problematic because Bayer was instituting a new branding strategy in preparation of the expiry of the aspirin patent in the United States. Thomas Edison, who needed phenol to manufacture phonograph
records, was also facing supply problems; in response, he created a
phenol factory capable of pumping out twelve tons per day. Edison's
excess phenol seemed destined for trinitrophenol production.
Although the United States remained officially neutral until
April 1917, it was increasingly throwing its support to the Allies
through trade. To counter this, German ambassador Johann Heinrich von Bernstorff and Interior Ministry official Heinrich Albert
were tasked with undermining American industry and maintaining public
support for Germany. One of their agents was a former Bayer employee,
Hugo Schweitzer.
Schweitzer set up a contract for a front company called the Chemical
Exchange Association to buy all of Edison's excess phenol. Much of the
phenol would go to the German-owned Chemische Fabrik von Heyden's
American subsidiary; Heyden was the supplier of Bayer's salicylic acid
for aspirin manufacture. By July 1915, Edison's plants were selling
about three tons of phenol per day to Schweitzer; Heyden's salicylic
acid production was soon back on line, and in turn Bayer's aspirin plant
was running as well.
The plot only lasted a few months. On 24 July 1915, Heinrich
Albert's briefcase, containing details about the phenol plot, was
recovered by a Secret Service
agent. Although the activities were not illegal—since the United States
was still officially neutral and still trading with Germany—the
documents were soon leaked to the New York World, an anti-German newspaper. The World published an exposé on 15 August 1915.
The public pressure soon forced Schweitzer and Edison to end the phenol
deal—with the embarrassed Edison subsequently sending his excess phenol
to the U.S. military—but by that time the deal had netted the plotters
over two million dollars and there was already enough phenol to keep
Bayer's Aspirin plant running. Bayer's reputation took a large hit,
however, just as the company was preparing to launch an advertising
campaign to secure the connection between aspirin and the Bayer brand.
Bayer loses foreign holdings
Beginning in 1915, Bayer set up a number of shell corporations
and subsidiaries in the United States, to hedge against the possibility
of losing control of its American assets if the U.S. should enter the
war and to allow Bayer to enter other markets (e.g., army uniforms).
After the U.S. declared war on Germany in April 1917, alien property custodianA. Mitchell Palmer
began investigating German-owned businesses, and soon turned his
attention to Bayer. To avoid having to surrender all profits and assets
to the government, Bayer's management shifted the stock to a new
company, nominally owned by Americans but controlled by the
German-American Bayer leaders. Palmer, however, soon uncovered this
scheme and seized all of Bayer's American holdings. After the Trading with the Enemy Act
was amended to allow sale of these holdings, the government auctioned
off the Rensselaer plant and all Bayer's American patents and
trademarks, including even the Bayer brand name and the Bayer cross
logo. It was bought by a patent medicine company, Sterling Products, Inc..
\The rights to Bayer Aspirin and the U.S. rights to the Bayer name and
trademarks were sold back to Bayer AG in 1994 for US$1 billion.
Interwar years
Bayer began advertising directly to American consumers just before the expiration of the aspirin patent. This ad, from The New York Times, 19 February 1917, emphasizes Bayer as the "One Real Aspirin" in anticipation of legal competition in the American market.
With the coming of the deadly Spanish flu
pandemic in 1918, aspirin—by whatever name—secured a reputation as one
of the most powerful and effective drugs in the pharmacopeia of the
time. Its fever-reducing properties gave many sick patients enough
strength to fight through the infection, and aspirin companies large and
small earned the loyalty of doctors and the public—when they could
manufacture or purchase enough aspirin to meet demand. Despite this,
some people believed that Germans put the Spanish flu bug in Bayer aspirin, causing the pandemic as a war tactic.
Newspaper
ad for Bayer Aspirin from April 1918. The aspirin patent had expired,
Bayer still had control over the Aspirin trademark, seen at the bottom
of the ad, and a "patriotic" slogan to buy war bonds. Also shows the
factory in New York State.
The U.S. ASA patent expired in 1917, but Sterling owned the aspirin trademark, which was the only commonly used term for the drug. In 1920, United Drug Company challenged the Aspirin
trademark, which became officially generic for public sale in the U.S.
(although it remained trademarked when sold to wholesalers and
pharmacists). With demand growing rapidly in the wake of the Spanish
flu, there were soon hundreds of "aspirin" brands on sale in the United
States.
Sterling Products, equipped with all of Bayer's U.S. intellectual
property, tried to take advantage of its new brand as quickly as
possible, before generic ASAs took over. However, without German
expertise to run the Rensselaer plant to make aspirin and the other
Bayer pharmaceuticals, they had only a finite aspirin supply and were
facing competition from other companies. Sterling president William E.
Weiss had ambitions to sell Bayer aspirin not only in the U.S., but to
compete with the German Bayer abroad as well. Taking advantage of the
losses Farbenfabriken Bayer (the German Bayer company) suffered through the reparation provisions of the Treaty of Versailles,
Weiss worked out a deal with Carl Duisberg to share profits in the
Americas, Australia, South Africa and Great Britain for most Bayer
drugs, in return for technical assistance in manufacturing the drugs.
Sterling also took over Bayer's Canadian assets as well as ownership of the Aspirin trademark which is still valid in Canada and most of the world. Bayer bought Sterling Winthrop
in 1994 restoring ownership of the Bayer name and Bayer cross trademark
in the US and Canada as well as ownership of the Aspirin trademark in
Canada.
Diversification of market
Aspro packaging 1931
Between World War I and World War II,
many new aspirin brands and aspirin-based products entered the market.
The Australian company Nicholas Proprietary Limited, through the
aggressive marketing strategies of George Davies, built Aspro into a global brand, with particular strength in Australia, New Zealand, and the U.K. American brands such as Burton's Aspirin, Molloy's Aspirin, Cal-Aspirin and St. Joseph Aspirin tried to compete with the American Bayer, while new products such Cafaspirin (aspirin with caffeine) and Alka-Seltzer (a soluble mix of aspirin and bicarbonate of soda) put aspirin to new uses. In 1925, the German Bayer became part of IG Farben, a conglomerate of former dye companies; IG Farben's brands of Aspirin and, in Latin America, the caffeinated Cafiaspirina (co-managed with Sterling Products) competed with less expensive aspirins such as Geniol.
Competition from new drugs
After World War II, with the IG Farben conglomerate dismantled because of its central role in the Nazi
regime, Sterling Products bought half of Bayer Ltd, the British Bayer
subsidiary—the other half of which it already owned. However, Bayer Aspirin made up only a small fraction of the British aspirin market because of competition from Aspro, Disprin
(a soluble aspirin drug) and other brands. Bayer Ltd began searching
for new pain relievers to compete more effectively. After several
moderately successful compound drugs that mainly utilized aspirin (Anadin and Excedrin),
Bayer Ltd's manager Laurie Spalton ordered an investigation of a
substance that scientists at Yale had, in 1946, found to be the
metabolically active derivative of acetanilide: acetaminophen. After clinical trials, Bayer Ltd brought acetaminophen to market as Panadol in 1956.
However, Sterling Products did not market Panadol in the United States or other countries where Bayer Aspirin still dominated the aspirin market. Other firms began selling acetaminophen drugs, most significantly, McNeil Laboratories with liquid Tylenol in 1955, and Tylenol pills in 1958. By 1967, Tylenol
was available without a prescription. Because it did not cause gastric
irritation, acetaminophen rapidly displaced much of aspirin's sales.
Another analgesic, anti-inflammatory drug was introduced in 1962: ibuprofen (sold as Brufen in the U.K. and Motrin
in the U.S.). By the 1970s, aspirin had a relatively small portion of
the pain reliever market, and in the 1980s sales decreased even more
when ibuprofen became available without prescription.
Also in the early 1980s, several studies suggested a link between children's consumption of aspirin and Reye's syndrome, a potentially fatal disease. By 1986, the U.S. Food and Drug Administration required warning labels on all aspirin, further suppressing sales. The makers of Tylenol also filed a lawsuit against Anacin aspirin maker American Home Products, claiming that the failure to add warning labels before 1986 had unfairly held back Tylenol sales, though this suit was eventually dismissed.
Investigating how aspirin works
The mechanism of aspirin's analgesic, anti-inflammatory and antipyretic
properties was unknown through the drug's heyday in the early- to
mid-twentieth century; Heinrich Dreser's explanation, widely accepted
since the drug was first brought to market, was that aspirin relieved
pain by acting on the central nervous system. In 1958 Harry Collier, a biochemist in the London laboratory of pharmaceutical company Parke-Davis, began investigating the relationship between kinins and the effects of aspirin. In tests on guinea pigs, Collier found that aspirin, if given beforehand, inhibited the bronchoconstriction effects of bradykinin. He found that cutting the guinea pigs' vagus nerve did not affect the action of bradykinin or
the inhibitory effect of aspirin—evidence that aspirin worked locally
to combat pain and inflammation, rather than on the central nervous
system. In 1963, Collier began working with University of London
pharmacology graduate student Priscilla Piper to determine the precise
mechanism of aspirin's effects. However, it was difficult to pin down
the precise biochemical goings-on in live research animals, and in vitro tests on removed animal tissues did not behave like in vivo tests.
After five years of collaboration, Collier arranged for Piper to work with pharmacologist John Vane at the Royal College of Surgeons of England, in order to learn Vane's new bioassay methods, which seemed like a possible solution to the in vitro testing failures. Vane and Piper tested the biochemical cascade associated with anaphylactic shock (in extracts from guinea pig lungs, applied to tissue from rabbitaortas).
They found that aspirin inhibited the release of an unidentified
chemical generated by guinea pig lungs, a chemical that caused rabbit
tissue to contract. By 1971, Vane identified the chemical (which they
called "rabbit-aorta contracting substance," or RCS) as a prostaglandin. In a 23 June 1971 paper in the journal Nature, Vane and Piper suggested that aspirin and similar drugs (the nonsteroidal anti-inflammatory drugs
or NSAIDs) worked by blocking the production of prostaglandins. Later
research showed that NSAIDs such as aspirin worked by inhibiting cyclooxygenase, the enzyme responsible for converting arachidonic acid into a prostaglandin.
Revival as heart drug
Aspirin's effects on blood clotting (as an antiplatelet agent) were first noticed in 1950 by Lawrence Craven. Craven, a family doctor in California, had been directing tonsillectomy patients to chew Aspergum, an aspirin-laced chewing gum.
He found that an unusual number of patients had to be hospitalized for
severe bleeding, and that those patients had been using very high
amounts of Aspergum. Craven began recommending daily aspirin to all his
patients, and claimed that the patients who followed the aspirin regimen
(about 8,000 people) had no signs of thrombosis. However, Craven's studies were not taken seriously by the medical community, because he had not done a placebo-controlled study and had published only in obscure journals.
The idea of using aspirin to prevent clotting diseases (such as
heart attacks and strokes) was revived in the 1960s, when medical
researcher Harvey Weiss found that aspirin had an anti-adhesive effect
on blood platelets (and unlike other potential antiplatelet drugs, aspirin had low toxicity). Medical Research Council
haematologist John O'Brien picked up on Weiss's finding and, in 1963,
began working with epidemiologist Peter Elwood on aspirin's
anti-thrombosis drug potential. Elwood began a large-scale trial of
aspirin as a preventive drug for heart attacks. Nicholas Laboratories
agreed to provide aspirin tablets, and Elwood enlisted heart attack
survivors in a double-blind
controlled study—heart attack survivors were statistically more likely
to suffer a second attack, greatly reducing the number of patients
necessary to reliably detect whether aspirin had an effect on heart
attacks. The study began in February 1971, though the researchers soon
had to break the double-blinding when a study by American epidemiologist
Hershel Jick suggested that aspirin prevented heart attacks but suggested that the heart attacks were more deadly.
Jick had found that fewer aspirin-takers were admitted to his hospital
for heart attacks than non-aspirin-takers, and one possible explanation
was that aspirin caused heart attack sufferers to die before reaching
the hospital; Elwood's initial results ruled out that explanation. When
the Elwood trial ended in 1973, it showed a modest but not statistically significant reduction in heart attacks among the group taking aspirin.
Several subsequent studies put aspirin's effectiveness as a heart
drug on firmer ground, but the evidence was not incontrovertible.
However, in the mid-1980s, with the relatively new technique of meta-analysis, statistician Richard Peto
convinced the U.S. FDA and much of the medical community that the
aspirin studies, in aggregate, showed aspirin's effectiveness with
relative certainty. By the end of the 1980s, aspirin was widely used as a preventive drug
for heart attacks and had regained its former position as the
top-selling analgesic in the U.S.
Affiliate Guest in Psychology, University of California, Santa Barbara
Why is my awareness here, while yours is over there? Why is the
universe split in two for each of us, into a subject and an infinity of
objects? How is each of us our own center of experience, receiving
information about the rest of the world out there? Why are some things conscious and others apparently not? Is a rat conscious? A gnat? A bacterium?
These questions are all aspects of the ancient “mind-body problem,”
which asks, essentially: What is the relationship between mind and
matter? It’s resisted a generally satisfying conclusion for thousands of
years.
Chalmers thought the mind-body problem should be called “hard” in
comparison to what, with tongue in cheek, he called the “easy” problems
of neuroscience: How do neurons and the brain work at the physical
level? Of course they’re not actually easy at all. But his point was
that they’re relatively easy compared to the truly difficult problem of
explaining how consciousness relates to matter.
Over the last decade, my colleague, University of California, Santa Barbara psychology professor Jonathan Schooler and I have developed what we call a “resonance theory of consciousness.”
We suggest that resonance – another word for synchronized vibrations –
is at the heart of not only human consciousness but also animal
consciousness and of physical reality more generally. It sounds like something the hippies might have dreamed up – it’s all vibrations, man! – but stick with me.
All about the vibrations
All things in our universe are constantly in motion, vibrating. Even
objects that appear to be stationary are in fact vibrating, oscillating,
resonating, at various frequencies. Resonance is a type of motion,
characterized by oscillation between two states. And ultimately all
matter is just vibrations of various underlying fields. As such, at every scale, all of nature vibrates.
Something interesting happens when different vibrating things come
together: They will often start, after a little while, to vibrate
together at the same frequency. They “sync up,” sometimes in ways that
can seem mysterious. This is described as the phenomenon of spontaneous self-organization.
Neuroscientists have identified sync in their research, too. Large-scale neuron firing occurs in human brains at measurable frequencies, with mammalian consciousness thought to be commonly associated with various kinds of neuronal sync.
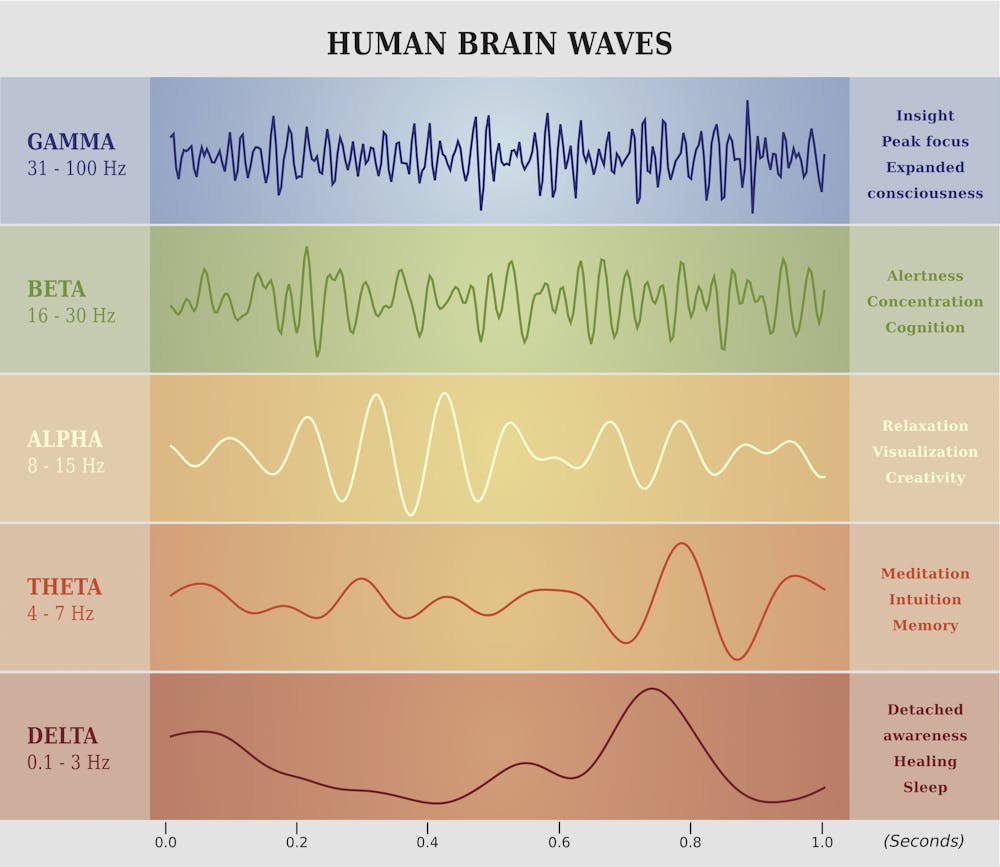
Fries focuses on gamma, beta and theta waves. These labels refer to
the speed of electrical oscillations in the brain, measured by
electrodes placed on the outside of the skull. Groups of neurons produce
these oscillations as they use electrochemical impulses to communicate
with each other. It’s the speed and voltage of these signals that, when
averaged, produce EEG waves that can be measured at signature cycles per
second.
Each type of synchronized activity is associated with certain types of brain function.artellia/Shutterstock.com
Gamma waves are associated with large-scale coordinated activities
like perception, meditation or focused consciousness; beta with maximum
brain activity or arousal; and theta with relaxation or daydreaming.
These three wave types work together to produce, or at least facilitate,
various types of human consciousness, according to Fries. But the exact
relationship between electrical brain waves and consciousness is still very much up for debate.
Fries calls his concept “communication through coherence.”
For him, it’s all about neuronal synchronization. Synchronization, in
terms of shared electrical oscillation rates, allows for smooth
communication between neurons and groups of neurons. Without this kind
of synchronized coherence, inputs arrive at random phases of the neuron
excitability cycle and are ineffective, or at least much less effective,
in communication.
A resonance theory of consciousness
Our resonance theory builds upon the work of Fries and many others,
with a broader approach that can help to explain not only human and
mammalian consciousness, but also consciousness more broadly.
Based on the observed behavior of the entities that surround us, from
electrons to atoms to molecules, to bacteria to mice, bats, rats, and
on, we suggest that all things may be viewed as at least a little
conscious. This sounds strange at first blush, but “panpsychism” – the
view that all matter has some associated consciousness – is an increasingly accepted position with respect to the nature of consciousness.
The panpsychist argues that consciousness did not emerge at some
point during evolution. Rather, it’s always associated with matter and
vice versa – they’re two sides of the same coin. But the large majority
of the mind associated with the various types of matter in our universe
is extremely rudimentary. An electron or an atom, for example, enjoys
just a tiny amount of consciousness. But as matter becomes more
interconnected and rich, so does the mind, and vice versa, according to
this way of thinking.
Biological organisms can quickly exchange information through various
biophysical pathways, both electrical and electrochemical.
Non-biological structures can only exchange information internally using
heat/thermal pathways – much slower and far less rich in information in
comparison. Living things leverage their speedier information flows
into larger-scale consciousness than what would occur in similar-size
things like boulders or piles of sand, for example. There’s much greater
internal connection and thus far more “going on” in biological
structures than in a boulder or a pile of sand.
Under our approach, boulders and piles of sand are “mere aggregates,”
just collections of highly rudimentary conscious entities at the atomic
or molecular level only. That’s in contrast to what happens in
biological life forms where the combinations of these micro-conscious
entities together create a higher level macro-conscious entity. For us,
this combination process is the hallmark of biological life.
The central thesis of our approach is this: the particular linkages
that allow for large-scale consciousness – like those humans and other
mammals enjoy – result from a shared resonance among many smaller
constituents. The speed of the resonant waves that are present is the
limiting factor that determines the size of each conscious entity in
each moment.
As a particular shared resonance expands to more and more
constituents, the new conscious entity that results from this resonance
and combination grows larger and more complex. So the shared resonance
in a human brain that achieves gamma synchrony, for example, includes a
far larger number of neurons and neuronal connections than is the case for beta or theta rhythms alone.
What about larger inter-organism resonance like a cloud of
fireflies with their little lights flashing in sync? Researchers think
their bioluminescent resonance arises due to internal biological oscillators that automatically result in each firefly syncing up with its neighbors.
Is the group of fireflies enjoying a higher level of group
consciousness? Probably not, since we can explain the phenomenon without
recourse to any intelligence or consciousness. But in biological
structures with the right kind of information pathways and processing
power, these tendencies toward self-organization can and often do
produce larger-scale conscious entities.
Our resonance theory of consciousness attempts to provide a unified
framework that includes neuroscience, as well as more fundamental
questions of neurobiology and biophysics, and also the philosophy of
mind. It gets to the heart of the differences that matter when it comes
to consciousness and the evolution of physical systems.
It is all about vibrations, but it’s also about the type of vibrations and, most importantly, about shared vibrations.
Sodium thiopental is an ultra-short-acting barbiturate that is marketed under the name Sodium Pentothal. It is often mistaken for "truth serum", or sodium amytal,
an intermediate-acting barbiturate that is used for sedation and to
treat insomnia, but was also used in so-called sodium amytal
"interviews" where the person being questioned would be much more likely
to provide the truth whilst under the influence of this drug. When
dissolved in water, sodium amytal can be swallowed, or it can be
administered by intravenous injection. The drug does not itself force
people to tell the truth, but is thought to decrease inhibitions and
slow creative thinking, making subjects more likely to be caught off
guard when questioned, and increasing the possibility of the subject
revealing information through emotional outbursts.
The memory-impairing effects and cognitive impairments induced by
sodium thiopental are thought to reduce a subject's ability to invent
and remember lies. This practice is no longer considered legally
admissible in court due to findings that subjects undergoing such
interrogations may form false memories, putting the reliability of all
information obtained through such methods into question. Nonetheless, it
is still employed in certain circumstances by defense and law
enforcement agencies as a "humane" alternative to torture interrogation
when the subject is believed to have information critical to the
security of the state or agency employing the tactic.
Chemistry
In 1988, the synthesis and binding studies of an artificial receptor binding barbiturates by six complementary hydrogen bonds was published. Since this first article, different kind of receptors were designed, as well as different barbiturates and cyanurates, not for their efficiencies as drugs but for applications in supramolecular chemistry, in the conception of materials and molecular devices.
Sodium barbital and barbital have also been used as pH buffers
for biological research, e.g., in immunoelectrophoresis or in fixative
solutions.
Side effects
Addiction
experts in psychiatry, chemistry, pharmacology, forensic science,
epidemiology, and the police and legal services engaged in delphic analysis
regarding 20 popular recreational drugs. Barbiturates were ranked 5th
in dependence, 3rd in physical harm, and 4th in social harm.
There are special risks to consider for older adults, women who are
pregnant, and babies. When a person ages, the body becomes less able to
rid itself of barbiturates. As a result, people over the age of
sixty-five are at higher risk of experiencing the harmful effects of
barbiturates, including drug dependence and accidental overdose.
When barbiturates are taken during pregnancy, the drug passes through
the placenta to the fetus. After the baby is born, it may experience
withdrawal symptoms and have trouble breathing. In addition, nursing
mothers who take barbiturates may transmit the drug to their babies
through breast milk. A rare adverse reaction to barbiturates is Stevens-Johnson syndrome, which primarily affects the mucous membranes.
Tolerance and dependence
With regular use, tolerance
to the effects of barbiturates develops. Research shows that tolerance
can develop with even one administration of a barbiturate. As with all
GABAergic drugs, barbiturate withdrawal produces potentially fatal
effects such as seizures in a manner reminiscent of delirium tremens and benzodiazepine withdrawal
although its more direct mechanism of GABA agonism makes barbiturate
withdrawal even more severe than that of alcohol or benzodiazepines
(subsequently making it one of the most dangerous withdrawals of any
known addictive substance). Similarly to benzodiazepines, the longer
acting barbiturates produce a less severe withdrawal syndrome than short
acting and ultra-short acting barbiturates. Withdrawal symptoms are
dose-dependent with heavier users being more affected than lower-dose
addicts.
The pharmacological treatment of barbiturate withdrawal is an
extended process often consisting of converting the patient to a
long-acting benzodiazepine (i.e. Valium),
followed by slowly tapering off the benzodiazepine. Mental cravings for
barbiturates can last for months or years in some cases and
counselling/support groups are highly encouraged by addiction
specialists. Patients should never try to tackle the task of
discontinuing barbiturates without consulting a doctor due to the high
lethality and relatively sudden onset of the withdrawal. Attempting to
quit "cold turkey" may result in serious neurological damage, severe
physical injuries received during convulsions, and even death via
glutamatergic excitotoxicity.
Overdose
Some symptoms of an overdose typically include sluggishness,
incoordination, difficulty in thinking, slowness of speech, faulty
judgement, drowsiness, shallow breathing, staggering, and, in severe
cases, coma or death. The lethal dosage of barbiturates varies greatly
with tolerance and from one individual to another. The lethal dose
is highly variable among different members of the class with
superpotent barbiturates such as pentobarbital being potentially fatal
in considerably lower doses than the low-potency barbiturates such as
butalbital. Even in inpatient settings, however, the development of
tolerance is still a problem, as dangerous and unpleasant withdrawal
symptoms can result when the drug is stopped after dependence has
developed. Tolerance to the anxiolytic and sedative effects of
barbiturates tends to develop faster than tolerance to their effects on
smooth muscle, respiration, and heart rate, making them generally
unsuitable for a long time psychiatric use. Tolerance to the
anticonvulsant effects tends to correlate more with tolerance to
physiological effects, however, meaning that they are still a viable
option for long-term epilepsy treatment.
Barbiturates in overdose with other CNS (central nervous system)
depressants (e.g. alcohol, opiates, benzodiazepines) are even more
dangerous due to additive CNS and respiratory depressant effects. In the
case of benzodiazepines, not only do they have additive effects,
barbiturates also increase the binding affinity of the benzodiazepine
binding site, leading to exaggerated benzodiazepine effects. (ex. If a
benzodiazepine increases the frequency of channel opening by 300%, and a
barbiturate increases the duration of their opening by 300%, then the
combined effects of the drugs increase the channels overall function by
900%, not 600%).
The longest-acting barbiturates have half-lives of a day or more, and subsequently result in bioaccumulation
of the drug in the system. The therapeutic and recreational effects of
long-acting barbiturates wear off significantly faster than the drug can
be eliminated, allowing the drug to reach toxic concentrations in the
blood following repeated administration (even when taken at the
therapeutic/prescribed dose) despite the user feeling little or no
effects from the plasma-bound concentrations of the drug. Users who
consume alcohol or other sedatives after the drugs effects have worn but
before it has cleared the system may experience a greatly exaggerated
effect from the other sedatives which can be incapacitating or even
fatal.
Barbiturates induce a number of hepatic CYP enzymes (most notably CYP2C9, CYP2C19 and CYP3A4), leading to exaggerated effects from many prodrugs
and decreased effects from drugs which are metabolized by these enzymes
to inactive metabolites. This can result in fatal overdoses from drugs
such as codeine, tramadol, and carisoprodol,
which become considerably more potent after being metabolized by CYP
enzymes. Although all known members of the class possess relevant enzyme
induction capabilities the degree of inhibition overall as well as the
impact on each specific enzyme span a broad range with phenobarbital and
secobarbital being the most potent enzyme inducers and butalbital and
talbutal being among the weakest enzyme inducers in the class.
Barbiturates act as positive allosteric modulators, and at higher doses, as agonists of GABAA receptors. GABA is the principal inhibitory neurotransmitter in the mammaliancentral nervous system (CNS). Barbiturates bind to the GABAA receptor at multiple homologous transmembrane pockets located at subunit interfaces, which are binding sites distinct from GABA itself and also distinct from the benzodiazepine
binding site. Like benzodiazepines, barbiturates potentiate the effect
of GABA at this receptor. In addition to this GABAergic effect,
barbiturates also block AMPA and kainate receptors, subtypes of ionotropic glutamate receptor.
Glutamate is the principal excitatory neurotransmitter in the mammalian
CNS. Taken together, the findings that barbiturates potentiate
inhibitory GABAA receptors and inhibit excitatory AMPA
receptors can explain the superior CNS-depressant effects of these
agents to alternative GABA potentiating agents such as benzodiazepines
and quinazolinones. At higher concentration, they inhibit the Ca2+-dependent release of neurotransmitters such as glutamate via an effect on P/Q-typevoltage-dependent calcium channels. Barbiturates produce their pharmacological effects by increasing the duration of chloride ion channel opening at the GABAA
receptor (pharmacodynamics: This increases the efficacy of GABA),
whereas benzodiazepines increase the frequency of the chloride ion
channel opening at the GABAA
receptor (pharmacodynamics: This increases the potency of GABA). The
direct gating or opening of the chloride ion channel is the reason for
the increased toxicity of barbiturates compared to benzodiazepines in overdose.
Further, barbiturates are relatively non-selective compounds that
bind to an entire superfamily of ligand-gated ion channels, of which
the GABAA receptor channel is only one of several representatives. This Cys-loop receptor superfamily of ion channels includes the neuronal nACh receptor channel, the 5-HT3 receptor channel, and the glycine receptor channel. However, while GABAA
receptor currents are increased by barbiturates (and other general
anaesthetics), ligand-gated ion channels that are predominantly
permeable for cationic ions are blocked by these compounds. For example,
neuronal nAChR channels are blocked by clinically relevant anaesthetic
concentrations of both thiopental and pentobarbital.
Such findings implicate (non-GABA-ergic) ligand-gated ion channels,
e.g. the neuronal nAChR channel, in mediating some of the (side) effects
of barbiturates. This is the mechanism responsible for the (mild to moderate) anesthetic
effect of barbiturates in high doses when used in anesthetic
concentration.
History
Barbituric acid was first synthesized November 27, 1864, by German chemist Adolf von Baeyer. This was done by condensingurea (an animal waste product) with diethyl malonate (an ester derived from the acid of apples).
There are several stories about how the substance got its name. The
most likely story is that Baeyer and his colleagues went to celebrate
their discovery in a tavern where the town's artillerygarrison were also celebrating the feast of Saint Barbara – the patron saint of artillerymen. An artillery officer is said to have christened the new substance by amalgamating Barbara with urea. Another story holds that Baeyer synthesized the substance from the collected urine of a Munich waitress named Barbara. No substance of medical value was discovered, however, until 1903 when two German scientists working at Bayer, Emil Fischer and Joseph von Mering, discovered that barbital was very effective in putting dogs to sleep. Barbital was then marketed by Bayer under the trade nameVeronal. It is said that Mering proposed this name because the most peaceful place he knew was the Italian city of Verona.
It was not until the 1950s that the behavioural disturbances and
physical dependence potential of barbiturates became recognized.
Barbituric acid itself does not have any direct effect on the central nervous system
and chemists have derived over 2,500 compounds from it that possess
pharmacologically active qualities. The broad class of barbiturates is
further broken down and classified according to speed of onset and
duration of action. Ultrashort-acting barbiturates are commonly used
for anesthesia
because their extremely short duration of action allows for greater
control. These properties allow doctors to rapidly put a patient
"under" in emergency surgery situations. Doctors can also bring a
patient out of anesthesia just as quickly, should complications arise
during surgery. The middle two classes of barbiturates are often
combined under the title "short/intermediate-acting." These
barbiturates are also employed for anesthetic purposes, and are also
sometimes prescribed for anxiety or insomnia.
This is not a common practice anymore, however, owing to the dangers
of long-term use of barbiturates; they have been replaced by the benzodiazepines
for these purposes. The final class of barbiturates are known as
long-acting barbiturates (the most notable one being phenobarbital,
which has a half-life of roughly 92 hours). This class of barbiturates
is used almost exclusively as anticonvulsants,
although on rare occasions they are prescribed for daytime sedation.
Barbiturates in this class are not used for insomnia, because, owing to
their extremely long half-life, patients would awake with a residual
"hang-over" effect and feel groggy.
Barbiturates can in most cases be used either as the free acid or
as salts of sodium, calcium, potassium, magnesium, lithium, etc. Codeine- and Dionine-based salts of barbituric acid have been developed. In 1912, Bayer introduced another barbituric acid derivative, phenobarbital, under the trade name Luminal, as a sedative-hypnotic.
Society and culture
Legal status
During World War II,
military personnel in the Pacific region were given "goofballs" to
allow them to tolerate the heat and humidity of daily working
conditions. Goofballs were distributed to reduce the demand on the
respiratory system, as well as maintaining blood pressure, to combat the
extreme conditions. Many soldiers returned with addictions that
required several months of rehabilitation before discharge. This led to
growing dependency problems, often exacerbated by indifferent doctors
prescribing high doses to unknowing patients through the 1950s and
1960s.
In the late 1950s and 1960s, an increasing number of published reports of barbiturate overdoses
and dependence problems led physicians to reduce their prescription,
particularly for spurious requests. This eventually led to the
scheduling of barbiturates as controlled drugs.
There is a small group of List II drugs for which doctors have to
write the prescriptions according to the same, tougher guidelines as
those for List I drugs (writing the prescription in full in letters,
listing the patients name, and have to contain the name and initials,
address, city and telephone number of the licensed prescriber issuing
the prescriptions, as well as the name and initials, address and city of
the person the prescription is issued to). Among that group of drugs
are the barbiturates amobarbital, butalbital, cyclobarbital, and pentobarbital.
In the United States, the Controlled Substances Act of 1970 classified most barbiturates as controlled substances—and they remain so as of September 2015. Barbital, methylphenobarbital (also known as mephobarbital), and phenobarbital are designated schedule IV
drugs, and "Any substance which contains any quantity of a derivative
of barbituric acid, or any salt of a derivative of barbituric acid" (all other barbiturates) were designated as being schedule III. Under the original CSA, no barbiturates were placed in schedule I, II, or V,
however amobarbital, pentobarbital, and secobarbital are schedule II
controlled substances unless they are in a suppository dosage form.
In 1971, the Convention on Psychotropic Substances was signed in Vienna. Designed to regulate amphetamines, barbiturates, and other synthetics, the 34th version of the treaty, as of 25 January 2014, regulates secobarbital as schedule II, amobarbital, butalbital, cyclobarbital, and pentobarbital as schedule III, and allobarbital, barbital, butobarbital, mephobarbital, phenobarbital, butabarbital, and vinylbital as schedule IV on its "Green List".[32] The combination medication Fioricet, consisting of butalbital, caffeine, and paracetamol (acetaminophen), however, is specifically exempted from controlled substance status, while its sibling Fiorinal, which contains aspirin instead of paracetamol and may contain codeine phosphate, remains a schedule III drug.
Recreational use
Recreational users report that a barbiturate high gives them feelings of relaxed contentment and euphoria. Physical and psychological dependence may also develop with repeated use.
Chronic misuse of barbiturates is associated with significant
morbidity. One study found that 11% of males and 23% of females with a sedative-hypnotic misuse die by suicide. Other effects of barbiturate intoxication include drowsiness, lateral and verticalnystagmus, slurred speech and ataxia,
decreased anxiety and loss of inhibitions. Barbiturates are also used
to alleviate the adverse or withdrawal effects of illicit drug use, in a
manner similar to long-acting benzodiazepines such as diazepam and clonazepam.
Often poly drug abuse occurs: Barbiturates are consumed with or
substituted by other available substances, most commonly alcohol.
Drug users tend to prefer short-acting and intermediate-acting barbiturates. The most commonly used are amobarbital (Amytal), pentobarbital (Nembutal), and secobarbital (Seconal). A combination of amobarbital and secobarbital (called Tuinal)
is also highly used. Short-acting and intermediate-acting barbiturates
are usually prescribed as sedatives and sleeping pills. These pills
begin acting fifteen to forty minutes after they are swallowed, and
their effects last from five to six hours.
Slang terms for barbiturates include barbs, bluebirds, dolls,
wallbangers, yellows, downers, goofballs, sleepers, 'reds & blues'
and tooties.
Examples
Generic structure of a barbiturate, including numbering scheme
Thiopental
is a barbiturate with one of the C-O double bonds (with the carbon
being labelled 2 in the adjacent diagram) replaced with a C-S double
bond, R1 being CH2CH3 and R2 being CH(CH3)CH2CH2CH3.