Nucleoside phosphoramidites are derivatives of natural or synthetic nucleosides. They are used to synthesizeoligonucleotides, relatively short fragments of nucleic acid and their analogs. Nucleoside phosphoramidites were first introduced in 1981 by Beaucage and Caruthers.
To avoid undesired side reactions, reactive hydroxy and exocyclic amino
groups present in natural or synthetic nucleosides are appropriately
protected. As long as a nucleoside analog contains at least one hydroxy
group, the use of the appropriate protecting strategy allows one to
convert that to the respective phosphoramidite and to incorporate the
latter into synthetic nucleic acids. To be incorporated in the middle of
an oligonucleotide chain using phosphoramidite strategy, the nucleoside
analog must possess two hydroxy groups or, less often, a hydroxy group
and another nucleophilic group (amino or mercapto). Examples include,
but are not limited to, alternative nucleotides, LNA, morpholino, nucleosides modified at the 2'-position (OMe, protected NH2, F), nucleosides containing non-canonical bases (hypoxanthine and xanthine contained in natural nucleosides inosine and xanthosine, respectively, tricyclic bases such as G-clamp, etc.) or bases derivatized with a fluorescent group or a linker arm.
Preparation
There are three main methods for the preparation of nucleoside phosphoramidites.
DMT = 4,4'-dimethoxytrityl; B = optionally protected nucleic base; R = phosphate protecting group
The common method involves treatment of a protected nucleoside bearing a
single free hydroxy group with phosphorodiamidite under the catalytic
action of a weak acid. Although some bisamidites were reported as thermally unstable compounds,
2-cyanoethyl N,N,N',N'-tetraisopropylphosphorodiamidite, the amidite
used to prepare commercial nucleoside phosphoramidites is relatively
stable. It can be synthesized using a two-step, one-pot procedure and
purified by vacuumdistillation.
An excellent review outlines the use of the latter reagent in
preparation of nucleosidic and non-nucleosidic phosphoramidites in great
detail.
In
the second method, the protected nucleoside is treated with the
phosphorochloridite in the presence of an organic base, most commonly N-ethyl-N,N-diisopropylamine (Hunig's base).
In the third method,
the protected nucleoside is first treated with chloro
N,N,N',N'-tetraisopropyl phosphorodiamidite in the presence of an
organic base, most commonly N-ethyl-N,N-diisopropylamine (Hunig's base)
to form a protected nucleoside diamidite. The latter is treated with an
alcohol respective to the desired phosphite protecting group, for
instance, 2-cyanoethanol, in the presence of a weak acid.
Nucleoside phosphoramidites are purified by column chromatography on silica gel.
To warrant the stability of the phosphoramidite moiety, it is advisable
to equilibrate the column with an eluent containing 3 to 5% of
triethylamine and maintain this concentration in the eluent throughout
the entire course of the separation. The purity of a phosphoramidite may
be assessed by 31P NMR spectroscopy.
As the P(III) atom in a nucleoside phosphoramidite is chiral, it
displays two peaks at about 149 ppm corresponding to the two
diastereomers of the compound. The potentially present phosphite
triester impurity displays peak at 138–140 ppm. H-phosphonate impurities
display peaks at 8 and 10 ppm.
Chemical properties of phosphoramidite moiety
Nucleoside
phosphoramidites are relatively stable compounds with a prolonged
shelf-life when stored as powders under anhydrous conditions in the
absence of air at temperatures below 4 °C. The amidites withstand mild
basic conditions. In contrast, in the presence of even mild acids,
phosphoramidites perish almost instantaneously. The phosphoramidites are
relatively stable to hydrolysis under neutral conditions. For instance,
half-life of 2-cyanoethyl 5'-O-(4,4'-dimethoxytrityl)thymidine-3'-O-(N,N-diisopropylamino)phosphite in 95% aqueous acetonitrile at 25 °C is 200 h.
X = O, S, NH.The
most important feature of phosphoramidites is their ability to undergo
the phosphoramidite coupling reaction that is, to react with
nucleophilic groups in the presence of an acidic azole catalyst, 1H-tetrazole, 2-ethylthiotetrazole, 2-benzylthiotetrazole, 4,5-dicyanoimidazole,
or a number of similar compounds. The reaction proceeds extremely
rapidly. This very feature makes nucleoside phosphoramidites useful
intermediates in oligonucleotide synthesis. Stereochemically, the phosphoramidite coupling leads to the epimerisation (forming of diastereomers) at the P(III) chiral center.
When water is served as a nucleophile, the product is an
H-phosphonate diester as shown in Scheme above. Due to the presence of
residual water in solvents and reagents, the formation of the latter
compound is the most common complication in the preparative use of
phosphoramidites, particularly in oligonucleotide synthesis.
X = S, Se.Phosphoramidites
are readily oxidized with weak oxidating reagents, for instance, with
aqueous iodine in the presence of weak bases or with hydrogen peroxide to form the respective phosphoramidates.
Similarly, phosphoramidites react with other chalcogens. When brought in contact with a solution of sulfur or a number of compounds collectively referred to as sulfurizing agents,phosphoramidites quantitatively form phosphorothioamidates. The reaction with selenium or selenium derivatives produces phosphoroselenoamidates. In all reactions of this type, the configuration at the phosphorus atom is retained.
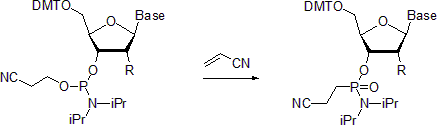
Nucleoside phosphoramidites undergo Michaelis-Arbuzov reaction to form the respective phosphonamidates. One example describes the preparation of phosphonamidates in the presence of acrylonitrile.
Reportedly, at room temperature the reaction is stereoselective with
the retention of configuration at the phosphorus center. In contrast,
when carried out at 55 °C, the reaction leads to racemized products.
Similarly to phosphines and tertiary phosphites, phosphoramidites readily undergo Staudinger reaction.
The
naturally occurring nucleotides (nucleoside-3'- or 5'-phosphates) and
their phosphodiester analogs are insufficiently reactive to afford an
expeditious synthetic preparation of oligonucleotides in high yields.
The selectivity and the rate of the formation of internucleosidic
linkages are dramatically improved by using 3'-O-(N,N-diisopropyl
phosphoramidite) derivatives of nucleosides (nucleoside
phosphoramidites) that serve as building blocks in phosphite triester
methodology. To prevent undesired side reactions, all other functional
groups present in nucleosides must be rendered unreactive (protected) by
attaching protecting groups.
Upon the completion of the oligonucleotide chain assembly, all the
protecting groups are removed to yield the desired oligonucleotides.
Below, the protecting groups currently used in commercially available and most common nucleoside phosphoramidite building blocks are briefly reviewed:
The 5'-hydroxyl group is protected by an acid-labile DMT (4,4'-dimethoxytrityl) group.
Thymine and uracil, nucleic bases of thymidine and uridine, respectively, do not have exocyclic amino groups and hence do not require any protection. In contrast, nucleic bases adenine, cytosine, and guanine
bear the exocyclic amino groups, which are reactive with the activated
phosphoramidites under the conditions of the coupling reaction.
Although, at the expense of additional steps in the synthetic cycle, the
oligonucleotide chain assembly may be carried out using
phosphoramidites with unprotected amino groups,
most often these are kept permanently protected over the entire length
of the oligonucleotide chain assembly. The protection of the exocyclic
amino groups must be orthogonal to that of the 5'-hydroxy group because
the latter is removed at the end of each synthetic cycle. The simplest
to implement and hence the most widely accepted is the strategy where
the exocyclic amino groups bear a base-labile protection. Most often,
two protection schemes are used.
In the first, the standard and more robust scheme (Figure), Bz (benzoyl) protection is used for A, dA, C, dC, G, and dG are protected with isobutyryl group. More recently, Ac (acetyl) group is often used to protect C and dC as shown in Figure.
In the second, mild protection scheme, A and dA are protected with isobutyryl or phenoxyacetyl groups (PAC). C and dC bear acetyl protection, and G and dG are protected with 4-isopropylphenoxyacetyl (i-Pr-PAC) or dimethylformamidino (dmf)
groups. Mild protecting groups are removed more readily than the
standard protecting groups. However, the phosphoramidites bearing these
groups are less stable when stored in solution.
The phosphite group is protected by a base-labile 2-cyanoethyl group.
Once a phosphoramidite has been coupled to the solid support-bound
oligonucleotide and the phosphite moieties have been converted to the
P(V) species, the presence of the phosphate protection is not mandatory
for the successful conducting of further coupling reactions.
2'-O-Protected ribonucleoside phosphoramidites.
In RNA synthesis, the 2'-hydroxy group is protected with TBDMS (t-butyldimethylsilyl) group. or with TOM (tri-iso-propylsilyloxymethyl) group,both being removable by treatment with fluoride ion.
The phosphite moiety also bears a diisopropylamino (iPr2N)
group reactive under acidic conditions. On activation, the
diisopropylamino group leaves, to be substituted by the 5'-hydroxy group
of the support-bound oligonucleotide.