From Wikipedia, the free encyclopedia
| Green Climate Fund | |
 |
|
| Abbreviation | GCF |
|---|---|
| Formation | 2010 |
| Legal status | Active |
| Headquarters | Songdo, Incheon, South Korea |
| Website | www |
The Green Climate Fund (GCF) is a fund within the framework of the UNFCCC founded as a mechanism to assist developing countries in adaptation and mitigation practices to counter climate change. The GCF is based in the new Songdo district of Incheon, South Korea. It is governed by a Board of 24 members and initially supported by a Secretariat.
‘The Green Climate Fund will support projects, programmes, policies and other activities in developing country Parties using thematic funding windows’.[1] It is intended to be the centrepiece of efforts to raise Climate Finance of $100 billion a year by 2020. This is not an official figure for the size of the Fund itself, however. Disputes also remain as to whether the funding target will be based on public sources, or whether "leveraged" private finance will be counted towards the total.[2] Only a fraction of this sum had been pledged as of July 2013, mostly to cover start-up costs.
According to the Climate & Development Knowledge Network, at the third meeting of the Board in Berlin, Germany, in March 2013, members agreed on how to move forward with the fund’s Business Model Framework (BMF). They identified the need to assess various options for how nations could access the fund, approaches for involving the private sector, plus ways to measure results and ensure requests for monies are country-driven.[3] At the fourth Board meeting in Songdo, South Korea, in June 2013, Hela Cheikhrouhou, a Tunisian national, was selected to become the Fund's first Executive Director.[4] "Resource mobilisation" (establishing a process for funding pledges) is expected to be the most contentious issue for the fifth Board meeting in Paris, France, in October 2013.[5]
History
The Copenhagen Accord, established during the 15th Conference Of the Parties (COP-15) in Copenhagen in 2009 mentioned the "Copenhagen Green Climate Fund". The fund was formally established during the 2010 United Nations Climate Change Conference in Cancun and is a fund within the UNFCCC framework.[6] Its governing instrument was adopted at the 2011 UN Climate Change Conference (COP 17) in Durban, South Africa.[7]Organization
During COP-16 in Cancun, the matter of governing the GCF was entrusted to the newly founded Green Climate Fund Board, and the World Bank was chosen as the temporary trustee.[6] To develop a design for the functioning of the GCF, the ‘Transitional Committee for the Green Climate Fund’ was established in Cancun too. The committee met four times throughout the year 2011, and submitted a report to the 17th COP in Durban, South Africa. Based on this report, the COP decided that the ‘GCF would become an operating entity of the financial mechanism’ of the UNFCCC,[8] and that on COP-18 in 2012, the necessary rules should be adopted to ensure that the GCF ‘is accountable to and functions under the guidance of the COP’.[8] Researchers at the Overseas Development Institute state that without this last minute agreement on a governing instrument for the GCF, the "African COP" would have been considered a failure.[9] Furthermore, the GCF Board was tasked with developing rules and procedures for the disbursement of funds, ensuring that these should be consistent with the national objectives of the countries where projects and programmes will be taking place. The GCF Board was also charged with establishing an independent secretariat and the permanent trustee of the GCF.[8]
Sources of finance
The Green Climate Fund is intended to be the centrepiece of Long Term Financing under the UNFCCC, which has set itself a goal of raising $100 billion per year by 2020. Uncertainty over where this money would come from led to the creation of a High Level Advisory Group on Climate Change Financing (AGF) was founded by UN Secretary-General Ban Ki-Moon in February 2010.There is no formal connection between this Panel and the GCF, although its report is one source for debates on "resource mobilisation" for the GCF, an item that will be discussed at the Fund's October 2013 Board meeting.[10]
The lack of pledged funds and potential reliance on the private sector is controversial and has been criticized by developing countries.[11]
Pledges to the fund reached $10.2 billion on May 28, 2015.[12]
Issues
The process of designing the GCF has raised several issues. These include ongoing questions on how funds will be raised,[13] the role of the private sector,[14] the level of "country ownership" of resources,[15] and the transparency of the Board itself.[16] In addition, questions have been raised about the need for yet another new international climate institution which may further fragment public dollars that are put toward mitigation and adaptation annually.[17]The Fund is also pledged to offer "balanced" support to adaptation and mitigation, although there is some concern amongst developing countries that inadequate adaptation financing will be offered, in particular if the fund is reliant on "leveraging" private sector finance.[18]
Role of the private sector
One of the most controversial aspects of the GCF concerns the creation of the Fund's Private Sector Facility (PSF). Many of the developed countries represented on the GCF board advocate a PSF that appeals to capital markets, in particular the pension funds and other institutional investors that control trillions of dollars that pass through Wall Street and other financial centers. They hope that the Fund will ultimately use a broad range of financial instruments.[19]However, several developing countries and non-governmental organizations have suggested that the PSF should focus on "pro-poor climate finance" that addresses the difficulties faced by micro-, small-, and medium-sized enterprises in developing countries. This emphasis on encouraging the domestic private sector is also written into the GCF’s Governing Instrument, its founding document.[20]
Additionality of funds
The Cancun agreements clearly specify that the funds provided to the developing countries as climate finance, including through the GCF, should be ‘new’ and ‘additional’ to existing development aid.[6]The condition of funds having to be new means that pledges should come on top of those made in previous years. As far as additionality is concerned, there is no strict definition of this term, which has already led to serious problems in evaluating the additionality of emission reductions through CDM-projects, leading to counterproductivity, and even fraud.[21][22]
A lack of stakeholder involvement
Using the money in the right way in order to enforce actual change on the ground is one of the biggest challenges ahead. Many academics argue that, in order to do this in an efficient way, all stakeholders should be involved in the process, instead of using a top-down approach. They point to that fact that, without their input, it is harder to achieve targets set. Moreover, projects often even miss out on their actual purpose.[18][23][24][25][26] A group of researchers associated with the Australian National University,[27] call for the foundation of so-called ‘National Implementing Entities’ (NIE) in each country, that would become responsible for ‘the implementation of sub-national projects’.[27] This would avoid national governments getting too involved, because in the past, they ‘often hindered the flow of international support to subnational scale reform for sustainable development’.[27] Overall, this view on the need for more stakeholder involvement can be framed within the movement in environmental governance calling for a shift from traditional ways of government to governance.[28] The Climate & Development Knowledge Network is funding a research project that aims to help the GCF Board, by analysing how best to allocate resources among countries. The project will research and present four case studies of how federal or central government money is presently distributed to sub-national entities. Chosen for the diversity in their underlying political systems, these are: China, India, Switzerland and the USA.[29]
Failure to ban fossil fuel funding under climate finance
At its board meeting in South Korea held in March 2015, the GCF refused an explicit ban on fossil fuel projects. Japan, China, and Saudi Arabia were opposing the ban. “It’s like a torture convention that doesn’t forbid torture,” Karen Orenstein, a campaigner for Friends of the Earth US who attended the meeting told the Guardian. “Honestly it should be a no-brainer at this point.”[30][31]
Accredited entities
- Acumen Fund, Inc. (US)
- Africa Finance Corporation (Nigeria)
- Agence Française de Développement (France)
- Asian Development Bank (Philippines)
- Caribbean Community Climate Change Center (Belize)
- Centre de Suivi Ecologique (Senégal)
- Conservation International Foundation (US)
- Corporcion Andina de Fomento (Venezuela)
- Deutsche Bank AktienGesellschaft (Germany)
- Environmental Investment Fund (Namibia)
- European Bank for Reconstruction and Development (United Kingdom)
- Inter-American Development Bank (US)
- International Bank for Reconstruction and Development and International Development Association (US)
- Kreditanstalt für Wiederaufbau (Germany)
- Ministry of Natural Resources (Rwanda)
- National Bank for Agriculture and Rural Development (India)
- Peruvian Trust Fund for National Parks and Protected Areas (Peru)
- Secretariat of the Pacific Regional Environment Programme (Samoa)
- United Nations Development Programme (US)
- United Nations Environment Programme (Kenya)
.jpg)
![\begin{align}
&U(z;R_{1},R_{2}) = -\frac{A}{6}\left(\frac{2R_{1}R_{2}}{z^2 - (R_{1} + R_{2})^2} + \frac{2R_{1}R_{2}}{z^2 - (R_{1} - R_{2})^2} + \ln\left[\frac{z^2-(R_{1}+ R_{2})^2}{z^2-(R_{1}- R_{2})^2}\right]\right)
\end{align}](http://upload.wikimedia.org/math/5/7/4/574d97354e30c09385b05957e1348e12.png)
 .
. , so that equation (1) for the potential energy function simplifies to:
, so that equation (1) for the potential energy function simplifies to:
 . This yields:
. This yields:












 is the center of mass of the molecule/group of particles.
is the center of mass of the molecule/group of particles.
 is the dipole operator and
is the dipole operator and  is the inversion operator. The permanent dipole moment of an atom in a non-degenerate state (see
is the inversion operator. The permanent dipole moment of an atom in a non-degenerate state (see 
 is an S-state, non-degenerate, wavefunction, which is symmetric or antisymmetric under inversion:
is an S-state, non-degenerate, wavefunction, which is symmetric or antisymmetric under inversion: 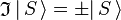 . Since the product of the wavefunction (in the ket) and its complex conjugate (in the bra) is always symmetric under inversion and its inverse,
. Since the product of the wavefunction (in the ket) and its complex conjugate (in the bra) is always symmetric under inversion and its inverse,
 , being a symmetry operator, is
, being a symmetry operator, is  and
and  may be moved from bra to ket and then becomes
may be moved from bra to ket and then becomes  . Since the only quantity that is equal to minus itself is the zero, the expectation value vanishes,
. Since the only quantity that is equal to minus itself is the zero, the expectation value vanishes,

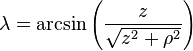


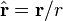 is the unit vector parallel to r;
is the unit vector parallel to r;

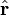 is a unit vector in the direction of r, p is the (vector)
is a unit vector in the direction of r, p is the (vector) 

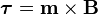
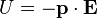 .
. .
.
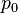 along the
along the 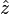 direction of the form
direction of the form
![\mathbf{E} = \frac{1}{4\pi\varepsilon_0} \left\{ \frac{\omega^2}{c^2 r}
( \hat{\mathbf{r}} \times \mathbf{p} ) \times \hat{\mathbf{r}}
+ \left( \frac{1}{r^3} - \frac{i\omega}{cr^2} \right) \left[ 3 \hat{\mathbf{r}} (\hat{\mathbf{r}} \cdot \mathbf{p}) - \mathbf{p} \right] \right\} e^{i\omega r/c} e^{-i\omega t}](http://upload.wikimedia.org/math/7/b/4/7b487096b3b9661fd46a5768a8a36407.png)

 , the far-field takes the simpler form of a radiating "spherical" wave, but with angular dependence embedded in the cross-product:
, the far-field takes the simpler form of a radiating "spherical" wave, but with angular dependence embedded in the cross-product: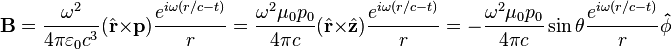

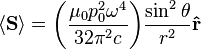
 ) responsible for such "donut-shaped" angular distribution is precisely the
) responsible for such "donut-shaped" angular distribution is precisely the  "p" wave.
"p" wave.




{kind=link}
{kind=link}