From Wikipedia, the free encyclopedia
https://en.wikipedia.org/wiki/Sulfur_isotope_biogeochemistry
Sulfur isotope biogeochemistry is the study of the distribution of sulfur isotopes in biological and geological materials. In addition to its common isotope, 32S, sulfur has three rare stable isotopes: 34S, 36S, and 33S. The distribution of these isotopes in the environment is controlled by many biochemical and physical processes, including biological metabolisms, mineral formation processes, and atmospheric chemistry. Measuring the abundance of sulfur stable isotopes in natural materials, like bacterial cultures, minerals, or seawater, can reveal information about these processes both in the modern environment and over Earth history.
Background
Natural abundance of sulfur isotopes

Sulfur has 24 known isotopes, 4 of which are stable (meaning that they do not undergo radioactive decay). 32S, the common isotope of sulfur, makes up 95.0% of the natural sulfur on Earth. In the atomic symbol of 32S, the number 32 refers to the mass of each sulfur atom in Daltons, the result of the 16 protons and 16 neutrons of 1 Dalton each that make up the sulfur nucleus. The three rare stable isotopes of sulfur are 34S (4.2% of natural sulfur), 33S (0.75%), and 36S (0.015%). These isotopes differ from 32S in the number of neutrons in each atom, but not the number of protons or electrons; as a result, each isotope has a slightly different mass, but has nearly identical chemical properties.
Physical chemistry
Small differences in mass between stable isotopes of the same element can lead to a phenomenon called an "isotope effect," where heavier or lighter isotopes are preferentially incorporated into different natural materials depending on the materials' chemical composition or physical state. Isotope effects are divided into two main groups: kinetic isotope effects and equilibrium isotope effects. A kinetic isotope effect occurs when a reaction is irreversible, meaning that the reaction only proceeds in the direction from reactants to products. Kinetic isotope effects cause isotopic fractionation—meaning that they affect the isotopic composition of reactant and product compounds—because the mass differences between stable isotopes can affect the rate of chemical reactions. It takes more energy to reach the transition state of a reaction if the compound has bonds with a heavier isotope, which causes the compound with heavier isotopes to react more slowly. Normal kinetic isotope effects cause the lighter isotope (or isotopes) to be preferentially included in a reaction's product. The products are then said to be "depleted" in the heavy isotope relative to the reactant. Rarely, inverse kinetic isotope effects may occur, where the heavier isotope is preferentially included in a reaction's product.
Equilibrium isotope effects cause fractionation because it is more chemically favorable for heavy isotopes to take part in stronger bonds. An equilibrium isotope effect occurs when a reaction is at equilibrium, meaning that the reaction is able to occur in both directions simultaneously. When a reaction is at equilibrium, heavy isotopes will preferentially accumulate where they can form the strongest bonds. For example, when the water in a sealed, half-full bottle is in equilibrium with the vapor above it, the heavier isotopes 2H and 18O will accumulate in the liquid, where they form stronger bonds, while the lighter isotopes 1H and 16O will accumulate in the vapor. The liquid is then said to be "enriched" in the heavy isotope relative to the vapor.
Calculations
Delta notation
Differences in the abundance of stable isotopes among natural materials are usually very small (natural differences in the ratio of rare to common isotope are almost always below 0.1%, and sometimes much smaller). Nevertheless, these very small differences can record meaningful biological and geological processes. To facilitate comparison of these small but meaningful differences, isotope abundances in natural materials are often reported relative to isotope abundances in designated standards. The convention for reporting the measured difference between a sample and a standard is called "delta notation." For example, imagine an element X for which we wish to compare the rare, heavy stable isotope with atomic mass A (AX) to the light, common isotope with atomic mass B (BX). The abundance of AX and BX in any given material is reported with the notation δAX. δAX for the sample material is calculated as follows:
AR = (total amount of AX)/(total amount of BX)
δAXsample = (ARsample - ARstandard)/ARstandard
δ values are most commonly reported in parts per thousand, commonly referred to in isotope chemistry as per mille and represented by the symbol ‰. To report δ values in per mille, the δ value as calculated above should be multiplied by 1000:
δAXsample (‰) = ((ARsample - ARstandard)/ARstandard) * 1000
Fractionation factors
While an isotope effect is the physical tendency for stable isotopes to distribute in a particular way, the isotopic fractionation is the measurable result of this tendency. The isotopic fractionation of a natural process can be calculated from measured isotope abundances. The calculated value is called a "fractionation factor," and allows the effect of different processes on isotope distributions to be mathematically compared. For example, imagine a chemical reaction Reactant → Product. Reactant and Product are materials that both contain the element X, and X has two stable isotopes, AX (the heavy isotope, with a mass of A) and BX (the light isotope, with a mass of B). The fractionation factor for the element X in the reaction Reactant → Product is represented by the notation AαProduct/Reactant. AαProduct/Reactant is calculated as follows:
AαProduct/Reactant = (δAXProduct + 1)/(δAXReactant + 1)
Fractionation factors can also be reported using the notation AεProduct/Reactant, which is sometimes called the "enrichment factor" and is calculated as follows:
AεProduct/Reactant = AαProduct/Reactant - 1
Like δ values, ε values can be reported in per mille by multiplying by 1000.
Δ33S and Δ36S notation
All kinetic and equilibrium isotope effects result from differences in atomic mass. As a result, a reaction that fractionates 34S will also fractionate 33S and 36S, and the fractionation factor for each isotope will be mathematically proportional to its mass. Because of the mathematical relationships of their masses, the observed relationships between δ34S, δ33S, and δ36S in most natural materials are approximately δ33S = 0.515 × δ34S and δ36S = 1.90 × δ34S. Rarely, natural processes can create deviations from this relationship, and these deviations are reported as Δ33S and Δ36S values, usually pronounced as "cap delta." These values are typically calculated as follows:
Δ33S = 1000 × [(1 + δ33S/1000) - (1 + δ34S 1000)0.518 - 1]
Δ36S = 1000 × [(1 + δ36S/1000) − (1 + δ34S/1000)1.91 − 1]
However, the method for calculating Δ33S and Δ36S values is not standardized, and can differ among publications.
.jpg)
Reference materials
Agreed-upon reference materials are required so that reported δ values are comparable among studies. For the sulfur isotope system, δ34S values are reported on the Vienna-Cañon Diablo Troilite (VCDT) scale. The original CDT scale was based on a sample of the mineral troilite recovered from the Canyon Diablo meteorite at Meteor Crater, Arizona, US. The Cañon Diablo Troilite was assigned a δ34S value of 0‰. However, troilite from the Canyon Diablo meteorite was later discovered to have variable sulfur isotope composition. As a result, VCDT was established as a hypothetical sulfur isotope reference with a 34R value of 0.044151 and δ34S of 0‰, but no physical sample of VCDT exists. Samples are now measured in comparison to International Atomic Energy Agency (IAEA) reference materials, which are well-characterized, lab-prepared compounds with known δ34S values. A commonly-used IAEA reference material is IAEA-S-1, a silver sulfide reference material with a δ34S value of -0.30‰ VCDT. 33S and 36S abundance can also be measured relative to IAEA reference materials and reported on the VCDT scale. For these isotopes, too, VCDT is established as having δ33S and δ36S values of 0‰. The 33R value of VCDT is 0.007877 and the 36R value is 0.0002. IAEA-S-1 has a 33R value of 0.0007878 and a δ33S value of -0.05‰ VCDT; it has a δ36S value of -0.6‰ VCDT.
Analytical methods and instrumentation
The sulfur isotopic composition of natural samples can be determined by Elemental Analysis-Isotope Ratio Mass Spectrometry (EA-IRMS), by Dual Inlet-Isotope Ratio Mass Spectrometry (DI-IRMS), by Multi-Collector-Inductively Coupled Plasma Mass Spectrometry (MC-ICPMS), by Secondary Ion Mass Spectrometry (SIMS), or by Nanoscale secondary ion mass spectrometry (NanoSIMS). MC-ICPMS can be paired with gas chromatography (GC-MC-ICPMS) to separate certain volatile compounds in a sample and measure the sulfur isotopic composition of individual compounds.
Natural variations in sulfur isotope abundance
Sulfur in natural materials

Sulfur is present in the environment in solids, gases, and aqueous species. Sulfur-containing solids on Earth include the common minerals pyrite (FeS2), galena (PbS), and gypsum (CaSO4•2H2O). Sulfur is also an important component of biological material, including in the essential amino acids cysteine and methionine, the B vitamins thiamine and biotin, and the ubiquitous substrate coenzyme A. In the ocean and other natural waters, sulfur is abundant as dissolved sulfate. Hydrogen sulfide is also present in some parts of the deep ocean where it is released from hydrothermal vents. Both sulfate and sulfide can be used by specialized microbes to obtain energy or to grow. Gases including sulfur dioxide and carbonyl sulfide make up the atmospheric component of the sulfur cycle. Any process that transports or chemically transforms sulfur between these many natural materials also has the potential to fractionate sulfur isotopes.
Sulfur isotopic abundance in natural materials

Sulfur in natural materials can vary widely in isotopic composition: compilations of the δ34S values of natural sulfur-containing materials include values ranging from -55‰ to 135‰ VCDT. The ranges of δ34S values vary across sulfur-containing materials: for example, the sulfur in animal tissue ranges from ~ -10 to +20‰ VCDT, while the sulfate in natural waters ranges from ~ -20 to +135‰ VCDT. The range of sulfur isotope abundances in different natural materials results from the isotope fractionation associated with natural processes like the formation and modification of those materials, discussed in the next section.
Processes that fractionate sulfur isotopes
Numerous natural processes are capable of fractionating sulfur isotopes. Microbes are capable of a wide variety of sulfur metabolisms, including the oxidation, reduction, and disproportionation (or simultaneous oxidation and reduction) of sulfur compounds. The effect of these metabolisms on sulfur isotopic composition of the reactants and products is also highly variable, depending on the rate of relevant reactions, availability of nutrients, and other biological and environmental parameters. As an example, the microbial reduction of sulfate to sulfide generally results in a 34S-depleted product, but the strength of this fractionation has been shown to range from 0 to 65.6‰ VCDT.
Many abiotic processes also fractionate sulfur isotopes. Small fractionations with ε values from 0-5‰ have been observed in the formation of the mineral gypsum, an evaporite mineral produced through the evaporation of seawater. Some sulfide minerals, including pyrite and galena, can form through thermochemical sulfate reduction, a process in which seawater sulfate trapped in seafloor rock is reduced to sulfide by geological heat as the rock is buried; this process generally fractionates sulfur more strongly than gypsum formation.
Prior to the rise of oxygen in Earth's atmosphere (referred to as the Great Oxidation Event), additional sulfur-fractionating processes referred to as mass-anomalous or mass-independent fractionation uniquely affected the abundance of 33S and 36S in the rock record. Mass-anomalous fractionations are rare, but they can occur through certain photochemical reactions of gases in the atmosphere. Studies have shown that photochemical reactions of atmospheric sulfur dioxide can cause substantial mass-anomalous fractionation of sulfur isotopes.
| Process | Range of observed 34ε (‰ VCDT) |
|---|---|
| Assimilatory sulfate reduction | -0.9 to -2.8 |
| Dissimilatory sulfate reduction | 0 to -65.6 |
| Sulfite reduction | +0.3 to -41 |
| Sulfide oxidation | +3 to -18.0 |
| Sulfur disproportionation | Sulfate: -0.6 to +20.2
Sulfide: -5.5 to -8.6 |
| Thermochemical sulfate reduction | +10 to +25 |
| Gypsum formation | 0 to +4.2 |
Biological processes that intake sulfur
All organisms metabolize sulfur, and it is incorporated into the structure of proteins, polysaccharides, steroids, and many coenzymes. The biological pathway by which an organism intakes and/or removes sulfur can have significant impacts on the sulfur isotope composition of the organism and its environment.

Some organisms take in relatively small amounts of sulfate in a process called assimilatory sulfate reduction, for the purpose of synthesizing compounds that contain sulfur, such as the amino acids methionine and cysteine that can then be used to make proteins. In phytoplankton, most of the sulfur taken in by this process is incorporated into biomass as proteins (~35%), sulfate esters (~20%), and low-weight sulfur-containing compounds (~40%). Literature on the isotopic fractionation effects of the assimilatory sulfate reduction pathway are noticeably less robust than those discussing other microbial sulfur pathways, but some sources believe there to be only very slight isotopic variations (δ34S = -4.4‰ to +0.5‰) in the resulting organic sulfur relative to the surrounding sulfate.

There is also another common pathway by which organisms intake sulfur. These microorganisms, which consume and reduce sulfate in relatively large quantities, are said to perform dissimilatory sulfate reduction. These organisms use sulfate reduction as an energy source as opposed to a way to synthesize new cell components, and remove the resulting sulfide as a waste product. Microbial sulfate reduction has been demonstrated to fractionate sulfur isotopes in bacteria, with some studies showing a dependence upon sulfate concentration and/or temperature. Studies examining dozens of species of dissimilatory sulfate reducing microbes have observed sulfur isotope fractionations ranging from -70‰ to +42.0‰.
While dissimilatory sulfate reduction and assimilatory sulfate reduction are two of the most common pathways by which organisms uptake and utilize sulfate, there are many other pathways by which living things intake sulfur. For example, sulfur oxidation of compounds like hydrogen sulfide and elemental sulfur is performed by lithotrophic bacteria and chemosynthetic archaea. Most animals obtain sulfur directly from the methionine and cysteine in the protein they consume.
Stable Isotopes in Plants
Methods of detection
Previous efforts to understand how sulfur metabolism and biosynthetic pathways relied on expensive labeling experiments using radioactive 35S. By leveraging natural assimilatory processes, stable isotopes ratios can be used to track what are the sources of sulfur for plants, what organs plants utilize in sulfur acquisition and how sulfur moves through plants.

Sulfur (S) stable isotope composition measurements are often done using an Elemental Analysis-Isotope Ratio Mass Spectrometer, (EA-IRMS) in which organic sulfur from biological samples is oxidized to sulfur dioxide (SO2) and analysed on a mass spectrometer. The gas is then analyzed for the ratio of the lighter (32S16O2) to the heavier (34S16O2) isotopologue and this ratio is then compared to sulfur isotope standards in order to calculate relative fractionations. In biological materials, sulfur is particularly scarce, making the abundance of S isotopes difficult to measure. The elemental S composition of plant matter is ≈0.2%, accounting for approximately 2 mmol/m2 in most leaf tissue. In order to reach detectable levels of 30 ng to 3 µg of elemental S to calculate reliable δ34S values, leaf tissue samples need to be between 2–5 mg.
Improvements in detection have been made in recent years in the utilization of gas chromatography coupled with multicollector ICP-MS (GC/MC-ICP-MS) to be able to measure pmol quantities of organic S.Additionally, ICP-MS has been used to measure nanomolar quantities of dissolved sulfate. Most studies have focused on measuring the bulk δ34S of plant tissues and few studies have been performed on measuring the δ34S of individual S-containing compounds. The coupling of high-performance liquid chromatography (HPLC) with ICP-MS has been proposed as a way to test individual S-containing compounds.
Sources
Each year, approximately 0.3 gigatons of elemental sulfur is converted into organic matter by photosynthetic organisms. This organic sulfur is allocated into a diversity of compounds such as amino acids – namely cysteine (Cys) and methionine (Met) – proteins, cofactors, antioxidants, sulfate groups, Fe-S centers and secondary metabolites. The three main sources of sulfur are atmospheric, soil, and aquatic.
Most vegetation can acquire sulfur from gaseous atmospheric compounds or various ions either in soil solutions or water bodies. Uptake of gaseous and dissolved sulfur compounds apparently occurs with little accompanying isotopic selectivity. Dissolved sulfate (SO42-) is considered to be the central pool which is metabolized by microorganisms and plants as most forms of atmospheric sulfur is oxidized into sulfate. Atmospheric sulfur is eventually returned to the soil when it is scrubbed from the atmosphere during precipitation or through dryfall.
Atmosphere
Many plants acquire sulfur through gaseous atmospheric compounds. Leaves of trees have δ34S values lying between those of air and soil, suggesting that there is uptake occurring from atmospheric and soil sources. The δ34S values of trees has also been demonstrated to be height dependent with the foliage at the tops of conifers, bull rushes and deciduous trees having δ34S values more reflective of the atmosphere and lower foliage having δ34S values closer to that of soil. It has been proposed that this is due to upper foliage exerting a canopy action on the lower branches, taking up atmospheric sulfur before it can reach lower levels. This is further supported with the epiphytic lichens and mosses having δ34S values close to atmospheric S compounds. This occurs due lichen and mosses having no access to soil and relying on the direct uptake of gaseous sulfur, dissolved sulfur through rainfall and dry fall accumulation, providing a cumulative record of atmospheric sulfur isotope composition.
Main forms of atmospheric sulfur come from the natural sulfur emissions formed biologically and emitted as H2S or organic sulfur gases such as DMS (dimethyl sulfide), COS (carbonyl sulfide), and CS2 (carbon disulfide). These gases are predominantly formed over oceans, wetlands, salt marshes, and estuaries by algae and bacteria. Anthropogenic emissions have increased the concentration of sulfur in the atmosphere mainly through emissions of SO2, from coal, oil, industrial processes, and biomass burning. In 2000, global anthropogenic emission of sulfur was estimated of 55.2-68 Tg S per year, which is much higher than the natural sulfur emissions estimated to be 34 Tg S per year. In the event of excess sulfur in plant tissue it has been demonstrated that when exposed to high doses of sulfur dioxide, plants emit hydrogen sulfide (H2S) and possibly other reduced sulfur compounds in response to high sulfur loading
Soil
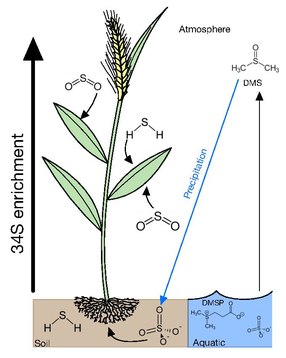
If soil sulfur is derived consistently from one source, the water-soluble and insoluble organic S fractions acquire similar isotopic compositions. In the case that there are two or more sources and/or if the isotopic composition of atmospheric or groundwater sulfate fluctuates, there may not be sufficient time for isotopic homogenization among the various forms of sulfur. The primary form of sulfur in soil is sulfate, which is transported upwards through the root system with minimal δ34S fractionation by 1-2‰. In contrast to higher canopy plants reflecting atmospheric δ34S, protected understory plants tend to reflect soil sulfur.
Aquatic
The forms of sulfur available in aquatic environments depends on whether it is a marine or freshwater environment. In marine environments, the main forms of sulfur available is in sulfate at ~29mM and a δ34S of 21‰ in the surface. This excess in sulfur is subsequently converted into dimethylsulfoniopropionate (DMSP) by algae as an osmolyte and a repellent against grazing. DMSP also accounts for 50-100% of bacterial sulfur demand making it the most important source of reduced sulfur for marine bacteria. DMSPs cleavage product dimethyl sulfide (DMS) is highly volatile escaping the ocean into the atmosphere with emissions ranging between 15 and 33 Tg S year−1 and accounting for 50-60% of the total natural reduced sulfur flux to the atmosphere.
Freshwater environments are more varied and depend on a multitude of factors, such as atmospheric deposition, runoff, diagenesis of bedrock and the presence of microbial sulfate reducers (MSR). Overall, the main sources of sulfur in freshwater environments are hydrogen sulfide and sulfate. In estuaries, plant roots extend into sulfide rich δ34S depleted sediments, created by MSR, and incorporate that into their biomass. Though levels of sulfide produced by MSR can be toxic and it has been proposed that these plants pump oxygen into their roots to oxidize sulfide into the less toxic sulfate. In these environments algaes will preferentially acquire the δ34S of HS− if present rather than the more abundant sulfate, as these sulfides can be readily incorporated into the direct formation of cysteine. This is consistent with cyanobacteria being able to carry out anoxygenic photosynthesis using sulfide.
Biochemistry
~90% of the organic-S in plants is concentrated in the amino acids cysteine and methionine. Cysteine acts as the direct or indirect precursor to any other organic-S compounds in plants such as coenzyme-A, methionine, biotin, lipoic acid and glutathione. The carbon skeleton necessary for S assimilation are provided by glycolysis (acetyl-CoA), respiration (aspartic acid, Asp, which derives from oxaloacetate) and photorespiration (serine, Ser). Because cysteine is a direct precursor to methionine, methionine is naturally 34S depleted in comparison to cysteine. The majority of sulfur is generally in the organic form but, when excess sulphur is available in the environment, inorganic sulfate becomes the major sulphur form. In most plants, 34S discrimination is minimal and in a study of rice plants it was observed that discrimination takes place in the uptake stage, depleting imported sulfate by 1-2‰ from the source. This effect is through the expression of SO42- transporter genes (SULTR), 14 of which have been identified – which are expressed dependent on the availability of sulfate in the environment. When sulfate is plentiful low affinity transporters are expressed and when sulfate is scarce high affinity genes with greater 34S discrimination.
Distribution through plant organs
Sulfate transported through the roots and SO2 diffusing into leaves becomes the pool for plants to assimilate sulfur throughout their tissues. Though there is minimal fractionation from the source sulfur of the total plant organic matter, in wheat, roots and stems are depleted from soil by 2‰ and leaves and grain are 2‰ enriched. The 34S enrichment in leaf whole matter is not caused by 34S-enriched sulfate present in the leaf, but is the result of the 34S-enrichment arriving at sink organs causing proteins in the leaves to be 34S-enriched. In rice, translocation from root to shoot does not discriminate S isotopes, however, the sulfate pools of the shoot are significantly 34S-enriched with respect to the sulfate pools of both root and sap. As sulfate, moves through the plant system and is incorporated into biomass, the pool becomes enriched, giving organs such as leaves and grains a higher δ34S than earlier tissues.
Applications
Rise of atmospheric oxygen
Signatures of mass-anomalous sulfur isotope fractionation preserved in the rock record have been an important piece of evidence for understanding the Great Oxidation Event, the sudden rise of oxygen on the ancient Earth. Nonzero values of Δ33S and Δ36S are present in the sulfur-bearing minerals of Precambrian rock formed greater than 2.45 billion years ago, but completely absent from rock less than 2.09 billion years old. Multiple mechanisms have been proposed for how oxygen prevents the fingerprints of mass-anomalous fractionation from being created and preserved; nevertheless, all studies of Δ33S and Δ36S records conclude that oxygen was essentially absent from Earth's atmosphere prior to 2.45 billion years ago.
Paleobiology and paleoclimate
A number of microbial metabolisms fractionate sulfur isotopes in distinctive ways, and the sulfur isotopic fingerprints of these metabolisms can be preserved in minerals and ancient organic matter. By measuring the sulfur isotopic composition of these preserved materials, scientists can reconstruct ancient biological processes and the environments where they occurred. δ34S values in the geologic record have been inferred to reveal the history of microbial sulfate reduction and sulfide oxidation. Paired δ34S and Δ33S records have also been used to show ancient microbial sulfur disproportionation.

Microbial dissimilatory sulfate reduction (MSR), an energy-yielding metabolism performed by bacteria in anoxic environments, is associated with an especially large fractionation factor. The observed 34εMSR values range from 0 to -65.6‰. Many factors influence the size of this fractionation, including sulfate reduction rate, sulfate concentration and transport, availability of electron donors and other nutrients, and physiological differences like protein expression. Sulfide produced through MSR may then go on to form the mineral pyrite, preserving the 34S-depleted fingerprint of MSR in sedimentary rocks. Many studies have investigated the δ34S values of ancient pyrite in order to understand past biological and environmental conditions. For example, pyrite δ34S records have been used to reconstruct shifts in primary productivity levels, changing ocean oxygen content, and glacial-interglacial changes in sea level and weathering. Some studies compare sulfur isotopes in pyrite to a second sulfur-containing material, like sulfate or preserved organic matter. Comparing pyrite to another material gives a fuller picture of how sulfur moved through ancient environments: it provides clues about the size of ancient 34εMSR values and the environmental conditions controlling MSR fractionation of sulfur isotopes.
Paleoceanography
δ34S records have been used to infer changes in seawater sulfate concentrations. Because the δ34S values of carbonate-associated sulfate are thought to be sensitive to seawater sulfate levels, these measurements have been used to reconstruct the history of seawater sulfate. δ34S values of pyrite have also been applied to reconstruct the concentration of seawater sulfate, based on expected biological fractionations at low sulfate concentrations. Both of these methods rely on assumptions about the depositional environment or the biological community, creating some uncertainty in the resulting reconstructions.


