| Ehlers–Danlos syndromes | |
|---|---|
.png) | |
| Individual with EDS displaying skin hyperelasticity | |
| Pronunciation |
|
| Specialty | Medical genetics, rheumatology |
| Symptoms | Overly flexible joints, stretchy skin, abnormal scar formation |
| Complications | Aortic dissection, joint dislocations, osteoarthritis |
| Usual onset | Birth or early childhood |
| Duration | Lifelong |
| Types | Hypermobile, classic, vascular, kyphoscoliosis, arthrochalasia, dermatosparaxis, brittle cornea syndrome, others |
| Causes | Genetic |
| Risk factors | Family history |
| Diagnostic method | Genetic testing, skin biopsy |
| Differential diagnosis | Marfan syndrome, cutis laxa syndrome, familial joint hypermobility syndrome |
| Treatment | Supportive |
| Prognosis | Depends on specific disorder |
| Frequency | 1 in 5,000 |
Ehlers–Danlos syndromes (EDS) are a group of genetic connective tissue disorders. Symptoms may include loose joints, joint pain, stretchy skin, and abnormal scar formation. These can be noticed at birth or in early childhood. Complications may include aortic dissection, joint dislocations, scoliosis, chronic pain, or early osteoarthritis.
EDS occurs due to variations of more than 19 different genes. The specific gene affected determines the type of EDS. Some cases result from a new variation occurring during early development, while others are inherited in an autosomal dominant or recessive manner. Typically, these variations result in defects in the structure or processing of the protein collagen. Diagnosis is often based on symptoms and confirmed with genetic testing or skin biopsy. However, people may initially be misdiagnosed with hypochondriasis, depression, or chronic fatigue syndrome.
There is no known cure. Treatment is supportive in nature. Physical therapy and bracing may help strengthen muscles and support joints. While some forms of EDS result in a normal life expectancy, those that affect blood vessels generally decrease life expectancy.
EDS affects at least one in 5,000 people globally. The prognosis depends on the specific disorder. Excess mobility was first described by Hippocrates in 400 BC. The syndromes are named after two physicians, Edvard Ehlers from Denmark and Henri-Alexandre Danlos from France, who described them at the turn of the 20th century.
Signs and symptoms
This
group of disorders affects connective tissues across the body, with
symptoms most typically present in the joints, skin, and blood vessels.
Effects may range from mildly loose joints to life-threatening cardiovascular complications. Due to the diversity of subtypes within the EDS family, symptoms may vary widely between individuals diagnosed with EDS.
Musculoskeletal
Musculoskeletal symptoms include hyperflexible joints that are unstable and prone to sprain, dislocation, subluxation, and hyperextension. There can be an early onset of advanced osteoarthritis, chronic degenerative joint disease, swan-neck deformity of the fingers, and Boutonniere deformity of the fingers. Tearing of tendons or muscles may occur. Deformities of the spine, such as scoliosis (curvature of the spine), kyphosis (a thoracic hump), tethered spinal cord syndrome, and occipitoatlantoaxial hypermobility may also be present. There can also be myalgia (muscle pain) and arthralgia (joint pain), which may be severe and disabling. Trendelenburg's sign is often seen, which means that when standing on one leg, the pelvis drops on the other side. Osgood–Schlatter disease, a painful lump on the knee, is common as well. In infants, walking can be delayed (beyond 18 months of age), and bottom-shuffling instead of crawling occurs.
 Individual with EDS showing hypermobile fingers, including the "swan-neck" malformation on the 2nd-5th digits, and a hypermobile thumb
Individual with EDS showing hypermobile fingers, including the "swan-neck" malformation on the 2nd-5th digits, and a hypermobile thumb
 Individual with EDS displaying hypermobile thumb
Individual with EDS displaying hypermobile thumb

 Kyphoscoliosis of the back of someone with kyphoscoliosis EDS.
Kyphoscoliosis of the back of someone with kyphoscoliosis EDS.




Skin
The weak connective tissue causes fragile skin that tears and bruises easily and atrophic scars that look like cigarette paper. Redundant skin folds occur, especially on the eyelids. Redundant skin folds are areas of excess skin lying in folds. Other skin symptoms include molluscoid pseudotumors, especially on pressure points, petechiae, subcutaneous spheroids, livedo reticularis, and piezogenic papules are less common. In vascular EDS, skin can also be thin and translucent. In dermatosparaxis EDS, the skin is extremely fragile and saggy.
 Atrophic scar in a case of EDS
Atrophic scar in a case of EDS
 Translucent skin in vascular EDS
Translucent skin in vascular EDS
 Individual with EDS displaying skin hyperelasticity
Individual with EDS displaying skin hyperelasticity
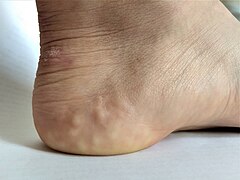 Piezogenic papules on the heel of an individual with hypermobile EDS
Piezogenic papules on the heel of an individual with hypermobile EDS

.png)


Cardiovascular
- Thoracic outlet syndrome
- Arterial rupture
- Valvular heart disease, such as mitral valve prolapse, creates an increased risk for infective endocarditis during surgery. This may progress to a life-threatening degree. Heart conduction abnormalities have been found in those with hypermobility form of EDS.
- Dilation and/or rupture (aneurysm) of ascending aorta
- Cardiovascular autonomic dysfunction such as postural orthostatic tachycardia syndrome
- Raynaud's phenomenon
- Varicose veins
- Heart murmur
- Heart conduction abnormalities
Other manifestations
- Hiatal hernia
- Gastroesophageal reflux
- Poor gastrointestinal motility
- Dysautonomia
- Gorlin's sign (touch tongue to nose)
- Anal prolapse
- Collapsed lung (spontaneous pneumothorax)
- Nerve disorders (carpal tunnel syndrome, acroparesthesia, neuropathy, including small fiber neuropathy)
- Insensitivity to local anesthetics.
- Arnold–Chiari malformation
- Platelet aggregation failure (platelets do not clump together properly)
- Mast cell disorders (including mast cell activation syndrome and mastocytosis)
- Pregnancy complications: increased pain, mild to moderate peripartum bleeding, cervical insufficiency, uterine tearing, or premature rupture of membranes.
- Hearing loss may occur in some types
- Eye: Nearsightedness, retinal tearing and retinal detachment, keratoconus, blue sclera, dry eye, Sjogren's syndrome, lens subluxation, angioid streaks, epicanthal folds, strabismus, corneal scarring, brittle cornea syndrome, cataracts, carotid-cavernous sinus fistulas, macular degeneration
- Cranial vertebral instability: caused by trauma(s) to the head and neck areas such as concussion and whiplash. Ligaments in neck are unable to heal properly, therefore, the neck structure does not have the ability to support the skull, which can then sink into the brain stem blocking the normal flow of cerebrospinal fluid, leading to issues related to the autonomic nervous system failing to work properly.
- Celiac disease may be associated with EDS. Also, it can be misdiagnosed as EDS due to common symptoms, including fatigue, pain, gastrointestinal complaints, or cardiovascular autonomic dysfunction.
 Gorlin's sign in a case of EDS.
Gorlin's sign in a case of EDS.
 An acute case of keratoconus in a case of Brittle cornea syndrome
An acute case of keratoconus in a case of Brittle cornea syndrome


Because it is often undiagnosed or misdiagnosed in childhood, some instances of EDS have been mischaracterized as child abuse.
The pain associated with the disorders may be severe.
Genetics

The
collagen fibril and EDS: (a) Normal collagen fibrils are of uniform
size and spacing. Fibrils from a person with dermatosparaxis (b) show
dramatic alterations in fibril morphology with severe effects on tensile
strength of connective tissues. Person with classical EDS (c) show
composite fibrils. Fibrils from a TNX-deficient person (d) are uniform
in size and no composite fibrils are seen. TNX-null (e) fibrils are less
densely packed and not as well aligned to neighboring fibrils.
Every type of EDS, except the hypermobile type, can be positively tied to specific genetic variation.
Variations in these genes can cause EDS:
- Collagen primary structure and collagen processing: ADAMTS2, COL1A1, COL1A2, COL3A1, COL5A1, COL5A2
- Collagen folding and collagen cross-linking: PLOD1, FKBP14
- Structure and function of myomatrix: TNXB, COL12A1
- Glycosaminoglycan biosynthesis: B4GALT7, B3GALT6, CHST14, DSE
- Complement pathway: C1R, C1S
- Intracellular processes: SLC39A13, ZNF469, PRDM5
Variations in these genes usually alter the structure, production, or processing of collagen or proteins
that interact with collagen. Collagen provides structure and strength
to connective tissue. A defect in collagen can weaken connective tissue
in the skin, bones, blood vessels, and organs, resulting in the features
of the disorder.
Inheritance patterns depend on the specific syndrome. Most forms of EDS
are inherited in an autosomal dominant pattern, which means only one of
the two copies of the gene in question must be altered to cause a
disorder. A few are inherited in an autosomal recessive pattern, which
means both copies of the gene must be altered for a person to be
affected by a disorder. It can also be an individual (de novo or "sporadic") variation. Sporadic variations occur without any inheritance.
Diagnosis
A diagnosis can be made by an evaluation of medical history and clinical observation. The Beighton criteria
are widely used to assess the degree of joint hypermobility. DNA and
biochemical studies can help identify affected individuals. Diagnostic
tests include collagen gene-variant testing, collagen typing via skin
biopsy, echocardiogram, and lysyl hydroxylase or oxidase activity.
However, these tests are not able to confirm all cases, especially in
instances of an unmapped variation, so clinical evaluation remains
important. If multiple individuals in a family are affected,
performing prenatal diagnosis may be possible using a DNA information
technique known as a linkage study. Knowledge about EDS among all kinds of practitioners is poor. Research is ongoing to identify genetic markers for all types.
Classification
In 2017, 13 subtypes of EDS were classified using specific diagnostic criteria.
According to the Ehlers-Danlos society, the syndromes can also be
grouped by the symptoms determined by specific gene mutations. Group A
disorders are those which affect primary collagen structure and
processing. Group B disorders affect collagen folding and crosslinking.
Group C are disorders of structure and function of myomatrix. Group D
disorders are those that affect glycosaminoglycan biosynthesis. Group E
disorders are characterized by defects in the complement pathway. Group F
are disorders of intracellular processes, and Group G is considered to
be unresolved forms of EDS.
Hypermobile EDS
Hypermobile
EDS (formerly categorized as type 3) is mainly characterized by
hypermobility that affects both large and small joints. It may lead to
frequent joint subluxations (partial dislocations) and dislocations. In
general, people with this variant have skin that is soft, smooth,
velvety, that bruises easily, and chronic muscle and/or bone pain. It affects the skin less than other forms. It has no available genetic test.
Hypermobility EDS (hEDS) is the most common of the 13 types of the
connective tissue disorder. Since there is no known genetic test,
providers have to diagnose hEDS based on what they already know about
the condition and the physical attributes that the patient shows. Other
than the general signs, attributes can include; faulty connective
tissues throughout the body, musculoskeletal issues and family history.
Along with these general signs and side effects, patients can have
trouble healing and problems with pregnancy.
Women that are pregnant should be warned about things like prelabor
rupture of membranes, drop in blood pressure with anesthesia,
precipitate birth (very fast active labor), malposition of bleeding, and
more. New mothers with hEDS should pay extra attention in taking care
of her new baby. Mothers may have trouble taking care of the baby
because of the risk of dropping the baby due to weak connective tissue
in arms and legs, falling, postpartum depression (more than the general
population), and healing from the birthing process.
Classical EDS
Classical
EDS (formerly categorized as type 1) is characterized by extremely
elastic skin that is fragile and bruises easily; and hypermobility of
the joints. Molluscoid pseudotumors (calcified hematomas that occur over
pressure points) and spheroids (cysts that contain fat occurring over
forearms and shins) also are seen often. A side complication of the
hyperelasticity presented in many cases of EDS makes it more difficult
for wounds to close on their own. Sometimes, motor development is delayed and hypotonia occurs. The variation causing this type of EDS is in the genes COL5A2, COL5A1, and less frequently COL1A1. It involves the skin more than hypermobile EDS.
In classical EDS there is often large variation in symptom presentation
from patient to patient. Because of this variance EDS has often been an
under diagnosed disorder.
Without genetic testing healthcare professionals may be able to provide
a provisional diagnosis based on careful examination of the mouth,
skin, and bones. As well as through neurological assessments.
The hyperelasticity of skin in EDS patients can be difficult to use in
diagnosis because there is no good standardized way to measure and
assess the elasticity of the skin. However, hyperelasticity is still a
good indicator as something that may point towards EDS along with other
symptoms. A good way to begin the diagnosis process is looking at family
history, EDS is an autosomal dominant condition and so is often
inherited from family members. Genetic testing remains the most reliable way for an EDS diagnosis to be made.
While there is no cure for type 1 EDS, a course of non weight bearing
exercising can help with muscular tension which can help correct some of
the symptoms of EDS. Anti inflammatory drugs as well as lifestyle
changes can help with joint pain. Lifestyle choices should also be made
with children that have EDS to try and prevent wounds to the skin.
Wearing protective garments can help with this. In the event of a wound
often deep stitches are used and left in place for a longer period of
time than normal.
Vascular variant of Ehlers–Danlos syndrome
Vascular EDS
(formerly categorized as type 4) is identified by skin that is thin,
translucent, extremely fragile, and bruises easily. It is also
characterized by fragile blood vessels and organs that can easily
rupture. Affected people are frequently short, and have thin scalp hair.
It also has characteristic facial features including large eyes, an
undersized chin, sunken cheeks, a thin nose and lips, and ears without
lobes.
Joint hypermobility is present, but generally confined to the small
joints (fingers, toes). Other common features include club foot, tendon
and/or muscle rupture, acrogeria (premature aging of the skin of the
hands and feet), early onset varicose veins, pneumothorax (collapse of a
lung), recession of the gums, and a decreased amount of fat under the
skin. It can be caused by the variations in the COL3A1 gene. Rarely, COL1A1 variations can also cause it.
Kyphoscoliosis EDS
Kyphoscoliosis
EDS (formerly categorized as type 6) is associated with severe
hypotonia at birth, delayed motor development, progressive scoliosis
(present from birth), and scleral fragility. People may also have easy
bruising, fragile arteries that are prone to rupture, unusually small
corneas, and osteopenia (low bone density). Other common features
include a "marfanoid habitus" which is characterized by long, slender
fingers (arachnodactyly), unusually long limbs, and a sunken chest
(pectus excavatum) or protruding chest (pectus carinatum). It can be caused by variations in the gene PLOD1, or rarely, in the FKBP14 gene.
Arthrochalasia EDS
Arthrochalasia
EDS (formerly categorized as types 7A & B) is characterized by
severe joint hypermobility and congenital hip dislocation. Other common
features include fragile, elastic skin with easy bruising, hypotonia,
kyphoscoliosis (kyphosis and scoliosis), and mild osteopenia.
Type-I collagen is usually affected. It is very rare, with about 30
cases reported. It is more severe than the hypermobility type.
Variations in the genes COL1A1 and COL1A2 cause it.
Dermatosparaxis EDS
Dermatosparaxis
EDS (formerly categorized as type 7C) is associated with extremely
fragile skin leading to severe bruising and scarring; saggy, redundant
skin, especially on the face; hypermobility ranging from mild to
serious; and hernias. Variations in the ADAMTS2 gene cause it. It is extremely rare, with around 11 cases reported.
Brittle cornea syndrome
Brittle cornea syndrome is characterized by the progressive thinning of the cornea, early-onset progressive keratoglobus or keratoconus, nearsightedness, hearing loss, and blue sclerae. Classic symptoms, such as hypermobile joints and hyperelastic skin, are also seen often. It has two types. Type 1 occurs due to variations in the ZNF469 gene. Type 2 is due to variations in the PRDM5 gene.
Classical-like EDS
Classical-like
EDS is characterized by skin hyperextensibility with velvety skin
texture and absence of atrophic scarring, generalized joint
hypermobility with or without recurrent dislocations (most often
shoulder and ankle), and easily bruised skin or spontaneous ecchymoses
(discolorations of the skin resulting from bleeding underneath). It can be caused by variations in the TNXB gene.
Spondylodysplastic EDS
Spondylodysplastic
EDS is characterized by short stature (progressive in childhood),
muscle hypotonia (ranging from severe congenital, to mild later-onset),
and bowing of limbs. It can be caused by variations in both copies of the B4GALT7 gene. Other cases can be caused by variations in the B3GALT6 gene.
People with variations in this gene can have kyphoscoliosis, tapered
fingers, osteoporosis, aortic aneurysma, and problems with the lungs.
Other cases can be caused by the SLC39A13 gene. Those with
variations in this gene have protuberant eyes, wrinkled palms of the
hands, tapering fingers, and distal joint hypermobility.
Musculocontractural EDS
Musculocontractural
EDS is characterized by congenital multiple contractures,
characteristically adduction-flexion contractures and/or talipes
equinovarus (clubfoot), characteristic craniofacial features, which are
evident at birth or in early infancy, and skin features such as skin
hyperextensibility, bruising, skin fragility with atrophic scars, and
increased palmar wrinkling. It can be caused by variations in the CHST14 gene. Some other cases can be caused by variations in the DSE gene.
Myopathic EDS
Myopathic
EDS (mEDS) is characterized by three major criteria: congenital muscle
hypotonia and/or muscle atrophy that improves with age, proximal joint
contractures of the knee, hip, and elbow, and hypermobility of distal
joints (ankles, wrists, feet, and hands).
There are also four minor criteria that may contribute to a diagnosis
of mEDS. This disorder can be inherited through either an autosomal
dominant or an autosomal recessive pattern. Molecular testing must be completed to verify that mutations in the COL12A1 gene are present; if not, other collagen-type myopathies should be considered.
Periodontal EDS
Periodontal EDS (pEDS) is an inherited autosomal dominant disorder
characterized by four major criteria of severe and intractable
periodontitis of early onset (childhood or adolescence), lack of
attached gingiva, pretibial plaques, and family history of a
first-degree relative who meets clinical criteria. Eight minor criteria may also contribute to the diagnosis of pEDS. Molecular testing may reveal mutations in C1R or C1S genes affecting the C1r protein.
Cardiac-valvular EDS
Cardiac-valvular
EDS (cvEDS) is characterized by three major criteria: severe
progressive cardiac-valvular problems (affecting aortic and mitral
valves), skin problems such as hyperextensibility, atrophic scarring,
thin skin, and easy bruising, and joint hypermobility (generalized or
restricted to small joints). There are also four minor criteria which may aid in diagnosis of cvEDS. Cardiac-valvular EDS is an autosomal recessive disorder, inherited through variation in both alleles of the gene COL1A2.
History
Until
1997, the classification system for EDS included 10 specific types, and
also acknowledged that other extremely rare types existed. At this time,
the classification system underwent an overhaul and was reduced to six
major types using descriptive titles. Genetic specialists recognize that
other types of this condition exist, but have only been documented in
single families. Except for hypermobility (type 3), the most common type of all ten types, some of the specific variations involved have been identified and they can be precisely identified by genetic testing;
this is valuable due to a great deal of variation in individual cases.
However, negative genetic test results do not rule out the diagnosis,
since not all of the variations have been discovered; therefore, the
clinical presentation is very important.
Forms of EDS in this category may present with soft, mildly
stretchable skin, shortened bones, chronic diarrhea, joint hypermobility
and dislocation, bladder rupture, or poor wound healing. Inheritance
patterns in this group include X-linked recessive, autosomal dominant,
and autosomal recessive. Examples of types of related syndromes other
than those above reported in the medical literature include:
- 305200 – type 5
- 130080 – type 8 – unspecified gene, locus 12p13
- 225310 – type 10 – unspecified gene, locus 2q34
- 608763 – Beasley–Cohen type
- 130070 – progeroid form – B4GALT7
- 130090 – type unspecified
- 601776 – D4ST1-deficient Ehlers–Danlos syndrome (adducted thumb-clubfoot syndrome) CHST14
Differential diagnosis
Several disorders share some characteristics with EDS. For example, in cutis laxa,
the skin is loose, hanging, and wrinkled. In EDS, the skin can be
pulled away from the body, but is elastic and returns to normal when let
go. In Marfan syndrome,
the joints are very mobile and similar cardiovascular complications
occur. People with EDS tend to have a "marfanoid" appearance (e.g.,
tall, skinny, long arms and legs, "spidery" fingers). However, physical
appearance and features in several types of EDS also have
characteristics including short stature, large eyes, and the appearance
of a small mouth and chin, due to a small palate. The palate can have a
high arch, causing dental crowding. Blood vessels can sometimes be
easily seen through translucent skin, especially on the chest. The
genetic connective tissue disorder, Loeys-Dietz syndrome, also has symptoms that overlap with EDS.
In the past, Menkes disease, a copper metabolism disorder, was thought to be a form of EDS. People are not uncommonly misdiagnosed with fibromyalgia, bleeding disorders,
or other disorders that can mimic EDS symptoms. Because of these
similar disorders and complications that can arise from an unmonitored
case of EDS, a correct diagnosis is important. Pseudoxanthoma elasticum (PXE) is worth consideration in diagnosis.
Management
There
is no known cure for Ehlers–Danlos syndromes and treatment is
supportive. Close monitoring of the cardiovascular system,
physiotherapy, occupational therapy, and orthopedic instruments (e.g.,
wheelchairs, bracing, casting) may be helpful. This can help stabilize
the joints and prevent injury. Orthopedic instruments are helpful for
the prevention of further joint damage, especially for long distances,
although individuals are advised not to become dependent on them until
other mobility options have been exhausted. People should avoid
activities that cause the joint to lock or overextend.
A physician may prescribe casting to stabilize joints. Physicians may refer a person to an orthotist for orthotic treatment
(bracing). Physicians may also consult a physical and/or occupational
therapist to help strengthen muscles and to teach people how to properly
use and preserve their joints.
Aquatic therapy promotes muscular development and coordination. With manual therapy, the joint is gently mobilized within the range of motion and/or manipulations.
If conservative therapy is not helpful, surgical joint repair may be
necessary. Medication to decrease pain or manage cardiac, digestive, or
other related conditions may be prescribed. To decrease bruising and improve wound healing, some people have responded to vitamin C.
Special precautions are often taken by medical care workers because of
the sheer number of complications that tend to arise in people with EDS.
In vascular EDS, signs of chest or abdominal pain are considered trauma
situations.
Cannabinoids and medical marijuana have shown some efficacy in reducing pain levels.
In general, medical intervention is limited to symptomatic
therapy. Before pregnancy, people with EDS should have genetic
counseling and familiarize themselves with the risks to their own bodies
that pregnancy poses. Children with EDS should be provided with
information about their disorder so they can understand why they should
avoid contact sports and other physically stressful activities. Children
should be taught that demonstrating the unusual positions that they can
maintain due to loose joints should not be done, as this may cause
early degeneration of the joints. Emotional support along with
behavioral and psychological therapy can be useful. Support groups can
be immensely helpful for people dealing with major lifestyle changes and
poor health. Family members, teachers, and friends should be informed
about EDS so they can accept and assist the child.
Surgery
The
instability of joints, leading to subluxations and joint pain, often
requires surgical intervention in people with EDS. Instability of almost
all joints can happen, but appears most often in the lower and upper
extremities, with the wrist, fingers, shoulder, knee, hip, and ankle
being most common.
Common surgical procedures are joint debridement, tendon replacements, capsulorrhaphy, and arthroplasty.
After surgery, the degree of stabilization, pain reduction, and
people's satisfaction can improve, but surgery does not guarantee an
optimal result: affected peoples and surgeons report being dissatisfied
with the results. Consensus is that conservative treatment is more
effective than surgery,
particularly since people have extra risks of surgical complications
due to the disease. Three basic surgical problems arise due to EDS: the
strength of the tissues is decreased, which makes the tissue less
suitable for surgery; the fragility of the blood vessels can cause
problems during surgery; and wound healing is often delayed or
incomplete.
If considering surgical intervention, seeking care from a surgeon with
extensive knowledge and experience in treating people with EDS and joint
hypermobility issues would be prudent.
Local anesthetics, arterial catheters, and central venous catheters cause a higher risk of bruise formation in people with EDS. Some people with EDS also show a resistance to local anaesthetics. Resistance to lidocaine and bupivacaine is not uncommon, and mepivacaine tends to work better in people with EDS. There are special recommendations for anesthesia in people with EDS. Detailed recommendations for anesthesia and perioperative care of people with EDS should be used to improve safety.
Surgery in people with EDS requires careful tissue handling and a longer immobilization afterward.
Prognosis
The
outcome for individuals with EDS depends on the specific type of EDS
they have. Symptoms vary in severity, even in the same disorder, and the
frequency of complications varies. Some people have negligible
symptoms, while others are severely restricted in daily life. Extreme
joint instability, chronic musculoskeletal pain, degenerative joint
disease, frequent injuries, and spinal deformities may limit mobility.
Severe spinal deformities may affect breathing. In the case of extreme
joint instability, dislocations may result from simple tasks such as
rolling over in bed or turning a doorknob. Secondary conditions such as
autonomic dysfunction or cardiovascular problems, occurring in any type,
can affect prognosis and quality of life. Severe mobility-related
disability is seen more often in hypermobile EDS than in classical EDS
or vascular EDS.
Although all types of EDS are potentially life-threatening, most
people have a normal lifespan. However, those with blood-vessel
fragility have a high risk of fatal complications, including spontaneous
arterial rupture, which is the most common cause of sudden death. The
median life expectancy in the population with vascular EDS is 48 years.
Epidemiology
Ehlers–Danlos
syndromes are inherited disorders estimated to occur in about one in
5,000 births worldwide. Initially, prevalence estimates ranged from one
in 250,000 to 500,000 people, but these estimates were soon found to be
too low, as more was studied about the disorders, and medical
professionals became more adept at diagnosis. EDS may be far more common
than the currently accepted estimate due to the wide range of
severities with which the disorder presents.
The prevalence of the disorders differs dramatically. The most
commonly occurring is hypermobile EDS, followed by classical EDS. The
others are very rare. For example, fewer than 10 infants and children
with dermatosparaxis EDS have been described worldwide.
Some types of EDS are more common in Ashkenazi Jews.
For example, the chance of being a carrier for dermatosparaxis EDS is
one in 248 in Ashkenazi Jews, whereas the prevalence of this variation
in the general population is one in 2,000.
Society and culture

Gary "Stretch" Turner showing his extreme Ehlers–Danlos syndrome
EDS may have contributed to the virtuoso violinist Niccolò Paganini's skill, as he was able to play wider fingerings than a typical violinist.
Many sideshow performers have EDS. Several of them were billed as
the Elastic Skin Man, the India Rubber Man, and Frog Boy. They included
such well-known individuals (in their time) as Felix Wehrle, James
Morris, and Avery Childs. Two performers with EDS currently hold world
records. Contortionist Daniel Browning Smith has hypermobile EDS and holds the current Guinness World Record for the most flexible man as of 2018, while Gary "Stretch" Turner
(shown right), sideshow performer in the Circus Of Horrors, has held
the current Guinness World Record for the most elastic skin since 1999,
for his ability to stretch the skin on his stomach 6.25 inches.
Notable cases

Pageant contestant Victoria Graham has EDS
- Actress Cherylee Houston, hypermobile EDS. She uses a wheelchair and was the first full-time disabled actress on Coronation Street.
- Drag queen Yvie Oddly
- Eric the Actor, a regular caller to the Howard Stern Show
- Actress and activist Jameela Jamil, hypermobile EDS. EDS is now part of her body positivity movements.
- Writer and actress Lena Dunham
- Australian singer Sia
- YouTuber and disability rights activist Annie Elainey
- Miss America pageant contestant (and Miss Allegany County) Victoria Graham


